Recognition: 2 theorem links
· Lean TheoremCrystal Fractional Graph Neural Network for Energy Prediction of High-Entropy Alloys
Pith reviewed 2026-05-12 01:03 UTC · model grok-4.3
The pith
A graph neural network fuses 16-atom local attention with global element fractions to predict high-entropy alloy energies at first-principles RMSE.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors claim that explicitly integrating local atomic interactions learned by graph attention layers on 16 on-site atoms with global compositional fractions through a feature fusion network produces total crystal energy predictions whose RMSE matches first-principles calculations and remains high even for low-energy configurations.
What carries the argument
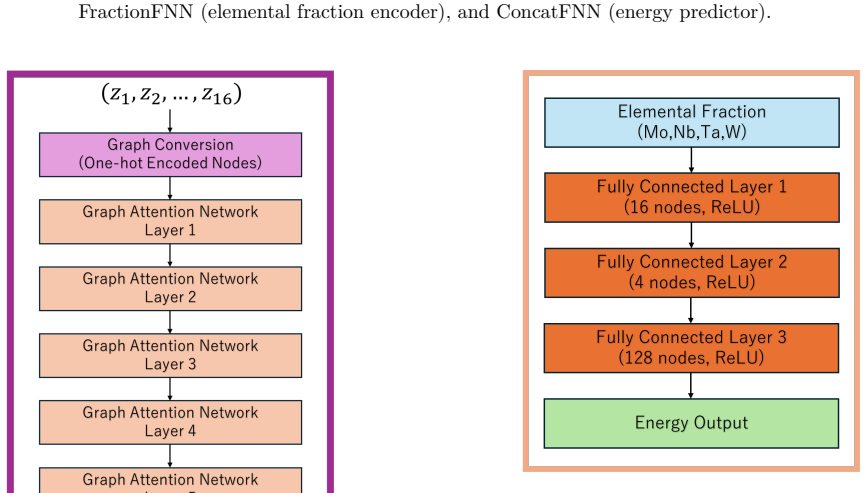
The feature fusion neural network that combines the output of the 16-atom graph attention network with the global fractional embedding network to yield the predicted crystal energy.
If this is right
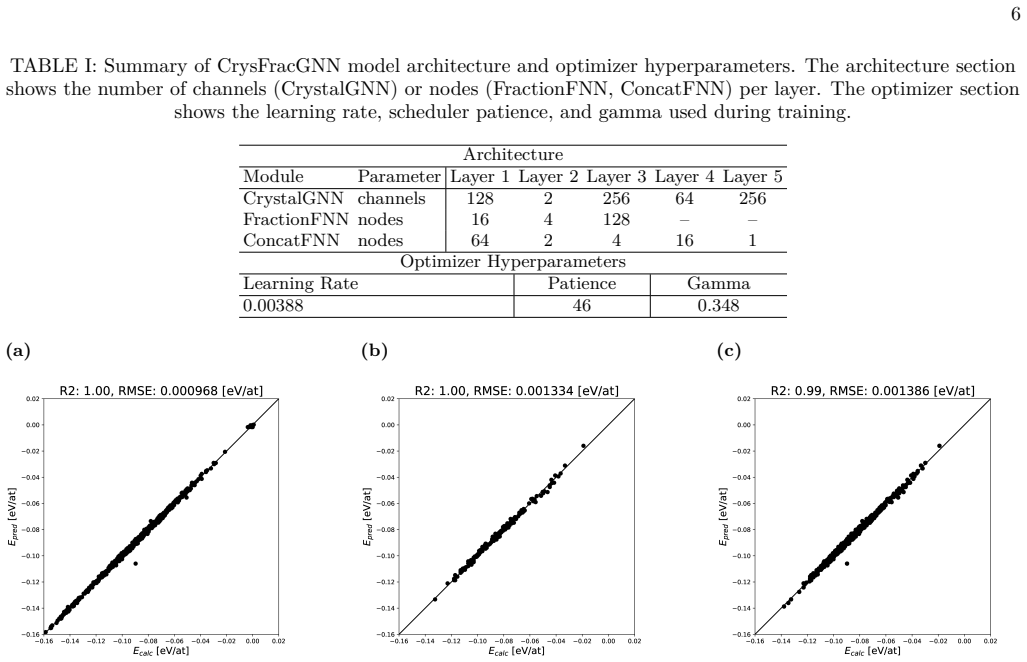
- The model achieves RMSE comparable to first-principles calculations on the validation set of 198 quaternary structures.
- High accuracy is maintained specifically on low-energy configurations.
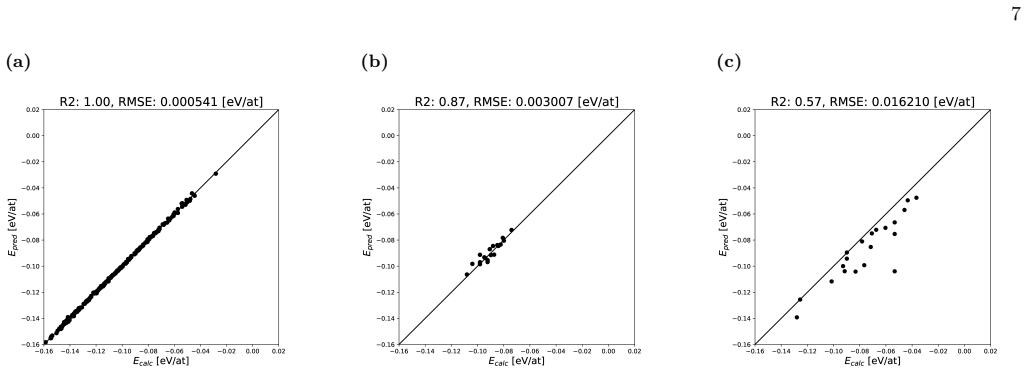
- The architecture is limited when applied to crystal cells larger than the 16-atom scale used in training.
- Hyperparameter optimization with Optuna produces the reported performance on the given dataset.
Where Pith is reading between the lines
- The fractional embedding component might be reusable as a drop-in module for other multi-principal-element materials beyond the quaternary HEAs tested here.
- Extending the local graph attention radius beyond 16 atoms could address the stated limitation on large cells without changing the global fusion step.
- If retrained on additional target properties such as formation enthalpy or elastic constants, the same local-plus-global architecture could support multi-property screening of HEAs.
- The approach suggests a general template for any crystal property where both short-range order and overall stoichiometry matter.
Load-bearing premise
That fusing the outputs of a fixed 16-atom graph attention network and a global fractional embedding network will yield reliable energy predictions that generalize to arbitrary high-entropy alloy compositions and cell sizes outside the 1,049 training structures.
What would settle it
First-principles energy calculations on a high-entropy alloy cell substantially larger than 16 atoms where the model's absolute error exceeds the reported RMSE would falsify the claim of reliable generalization.
Figures





read the original abstract
High-entropy alloys (HEAs) have attracted growing attention for their exceptional mechanical and thermal properties arising from complex atomic configurations. In this paper, we propose crystal fractional graph neural network for predicting the energy of high-entropy alloys by explicitly integrating both local atomic environments and global compositional information. The model consists of three components: a crystal graph neural network, which employs graph attention network layers to learn local interactions among 16 on-site atoms within the crystal lattice; fractional neural network, a fully connected network that embeds the global fraction of constituent elements; and feature fusion neural network, which fuses the outputs of the two submodels to predict the total crystal energy. We train the model on a dataset of 1,049 crystal structures and validate it on 198 quaternary structures, optimizing all hyperparameters via Optuna. Our results show that our model achieves an RMSE comparable to first-principles calculations and maintains high accuracy even for low-energy configurations. However, the model exhibits limitations in handling large crystal cells, which we aim to address in future work to extend its applicability to more complex systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a Crystal Fractional Graph Neural Network (CF-GNN) for energy prediction in high-entropy alloys. It combines a graph attention network operating on local 16-atom crystal graphs, a fully connected fractional embedding network for global elemental fractions, and a fusion network to output total crystal energy. The model is trained on 1,049 structures and validated on 198 quaternary structures with hyperparameters tuned by Optuna; the abstract claims RMSE performance comparable to first-principles calculations and good accuracy on low-energy configurations, while noting limitations for large crystal cells.
Significance. If the performance claims are supported by explicit numerical benchmarks against DFT and other ML baselines, and if the architecture can be extended beyond the fixed 16-atom regime, the work could provide a useful surrogate model for screening HEA configurational energies. The explicit separation of local atomic interactions and global composition is a sensible inductive bias for compositionally disordered systems. However, the current fixed-size design and absence of demonstrated extrapolation limit its immediate impact on the broader HEA literature, which routinely employs larger supercells.
major comments (3)
- [Abstract] Abstract: the claim that the model 'achieves an RMSE comparable to first-principles calculations' supplies no numerical RMSE value, error bars, baseline comparisons, or details on how low-energy configurations were identified or evaluated. This absence prevents verification of the central performance assertion.
- [Abstract] Abstract and model description: the local component is built exclusively around graph attention layers on exactly 16 on-site atoms per crystal graph, with training and validation restricted to 1,049 + 198 structures that fit this size. High-entropy alloy energy predictions for arbitrary compositions routinely require 32–128 atom supercells to sample configurational disorder; the fusion mechanism provides no size-invariant representation or extrapolation path, directly contradicting the claimed applicability.
- [Abstract] Validation protocol: the 198 quaternary validation structures are described only as 'quaternary structures' without clarification on whether they constitute true extrapolation to unseen compositions or cell sizes, or merely interpolation within the same 16-atom regime used for training.
minor comments (2)
- The abstract refers to 'crystal fractional graph neural network' without defining the acronym CF-GNN or providing a clear equation for the fusion step; this should be introduced with a schematic in the methods section.
- No mention is made of the specific loss function, optimizer, or convergence criteria used during Optuna tuning; these details are needed for reproducibility.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript describing the Crystal Fractional Graph Neural Network (CF-GNN). We address each major comment point by point below, indicating revisions where the manuscript will be updated to improve clarity and precision.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that the model 'achieves an RMSE comparable to first-principles calculations' supplies no numerical RMSE value, error bars, baseline comparisons, or details on how low-energy configurations were identified or evaluated. This absence prevents verification of the central performance assertion.
Authors: We agree that the abstract would benefit from explicit numerical support for the performance claim. The main text reports the specific RMSE value achieved by CF-GNN along with comparisons to DFT and details on low-energy configuration selection. In the revised manuscript we will incorporate these quantitative results, including the RMSE figure and error characterization, directly into the abstract to enable immediate verification. revision: yes
-
Referee: [Abstract] Abstract and model description: the local component is built exclusively around graph attention layers on exactly 16 on-site atoms per crystal graph, with training and validation restricted to 1,049 + 198 structures that fit this size. High-entropy alloy energy predictions for arbitrary compositions routinely require 32–128 atom supercells to sample configurational disorder; the fusion mechanism provides no size-invariant representation or extrapolation path, directly contradicting the claimed applicability.
Authors: The manuscript already states that the current implementation is restricted to 16-atom cells and explicitly notes limitations for larger cells, with future work planned to address this. The fusion mechanism combines local 16-atom graph attention with global composition fractions but does not claim size invariance or extrapolation beyond the trained regime. We will revise the abstract and model description to more precisely delimit the scope to 16-atom structures, removing any implication of immediate applicability to 32–128 atom supercells while retaining the stated future extension plans. revision: partial
-
Referee: [Abstract] Validation protocol: the 198 quaternary validation structures are described only as 'quaternary structures' without clarification on whether they constitute true extrapolation to unseen compositions or cell sizes, or merely interpolation within the same 16-atom regime used for training.
Authors: We will add explicit clarification in the revised methods, results, and abstract sections. The 198 quaternary structures employ elemental compositions absent from the training set (extrapolation in composition) while using the identical 16-atom cell size (interpolation in cell size). This distinction will be stated clearly to accurately characterize the validation protocol. revision: yes
Circularity Check
No circularity: standard supervised ML training and held-out validation
full rationale
The paper defines a fixed architecture (graph attention on exactly 16 atoms + fractional embedding + fusion network), trains it on 1,049 structures, validates on 198 held-out quaternary structures, and reports RMSE after Optuna hyperparameter search. This is ordinary empirical model evaluation against external DFT-derived labels; no equation, prediction, or uniqueness claim reduces to its own inputs by construction. The paper explicitly notes the 16-atom limitation and does not present any self-citation, ansatz smuggling, or fitted parameter as an independent first-principles result. The derivation chain is therefore self-contained and non-circular.
Axiom & Free-Parameter Ledger
free parameters (2)
- Neural network weights and biases
- Hyperparameters
axioms (2)
- domain assumption Graph attention networks can learn meaningful local atomic interactions from crystal graphs limited to 16 on-site atoms.
- domain assumption A fully connected network can embed global elemental fractions in a way that complements local structural features for energy prediction.
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/ArithmeticFromLogic.leanLogicNat recovery and J-cost uniqueness unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
crystal graph neural network, which employs graph attention network layers to learn local interactions among 16 on-site atoms within the crystal lattice; fractional neural network, a fully connected network that embeds the global fraction of constituent elements
-
IndisputableMonolith/Foundation/AlexanderDuality.leanalexander_duality_circle_linking (D=3 forcing) unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
the model exhibits limitations in handling large crystal cells
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
The GAT layer proposed by Veličković et al
CrystalGNN Architecture CrystalGNN has several Graph Attention Network (GAT) layers and each layer has a ReLU activation func- tion. The GAT layer proposed by Veličković et al. makes the GNN layer have different weights for each node [ 35]. For applying GNN layers, we convert 16 features to graph data which has 16 nodes and input this data to the GNN mode...
-
[2]
FractionFNN Architecture As shown in Fig. 5, FractionFNN is a fully connected neural network consisting of several layers, each fol- lowed by a ReLU activation function. The input to this network is the four-dimensional composition vec- tor (xMo, xNb, xTa, xW), where each component is in the range [0, 1] and is computed as the fractional occurrence of the...
-
[3]
ConcatFNN Architecture As illustrated in Fig. 6, ConcatFNN is a fully con- nected neural network consisting of several layers, each followed by a ReLU activation function. It takes two inputs: the output of CrystalGNN and the output of FractionFNN. To convert the CrystalGNN output, which has node-wise features, into a fixed-size vector suitable for Concat...
-
[4]
J. Yeh, S. Chen, S. Lin, J. Gan, T. Chin, T. Shun, C. Tsau, and S. Chang, Nanostructured high-entropy al- loys with multiple principal elements: Novel alloy design concepts and outcomes, Advanced Engineering Materials 7 (a) (b) (c) FIG. 8: The yyplot of the best model trained by 1247-point datasets for (a) low-energy test data, (b) 54-atom test data, and ...
work page 2004
- [5]
- [6]
-
[7]
M.-H. Tsai and J.-W. Yeh, High-entropy alloys: A critical review, Materials Research Letters 2, 107 (2014)
work page 2014
-
[8]
Y. Ye, Q. Wang, J. Lu, C. Liu, and Y. Yang, High- entropy alloy: challenges and prospects, Materials Today 19, 349 (2016)
work page 2016
-
[9]
C. Lu, L. Niu, N. Chen, K. Jin, T. Yang, P. Xiu, Y. Zhang, F. Gao, H. Bei, S. Shi, M.-R. He, I. M. Robert- son, W. J. Weber, and L. Wang, Enhancing radiation tolerance by controlling defect mobility and migration pathways in multicomponent single-phase alloys, Nature Communications 7, 13564 (2016)
work page 2016
-
[10]
E. P. George, D. Raabe, and R. O. Ritchie, High-entropy alloys, Nature Reviews Materials 4, 515 (2019)
work page 2019
-
[11]
O. El-Atwani, N. Li, M. Li, A. Devaraj, J. K. S. Baldwin, M. M. Schneider, D. Sobieraj, J. S. Wróbel, D. Nguyen- Manh, S. A. Maloy, and E. Martinez, Outstanding ra- diation resistance of tungsten-based high-entropy alloys, Science Advances 5, eaav2002 (2019)
work page 2019
-
[12]
Q. Xu, H. Q. Guan, Z. H. Zhong, S. S. Huang, and J. J. Zhao, Irradiation resistance mechanism of the cocrfemnni equiatomic high-entropy alloy, Scientific Reports 11, 608 (2021)
work page 2021
-
[13]
M. Dada, P. Popoola, S. Adeosun, and N. Mathe, High entropy alloys for aerospace applications, in Aerodynam- ics, edited by M. Gorji-Bandpy and A.-M. Aly (Inte- chOpen, Rijeka, 2019) Chap. 7
work page 2019
-
[14]
J. Dabrowa, W. Kucza, G. Cieślak, T. Kulik, M. Danielewski, and J.-W. Yeh, Interdiffusion in the fcc- structured al-co-cr-fe-ni high entropy alloys: Experimen- tal studies and numerical simulations, Journal of Alloys and Compounds 674, 455 (2016)
work page 2016
-
[15]
I. Ondicho, M. Choi, W.-M. Choi, J. B. Jeon, H. R. Ja- farian, B.-J. Lee, S. I. Hong, and N. Park, Experimen- tal investigation and phase diagram of cocrmnni–fe sys- tem bridging high-entropy alloys and high-alloyed steels, Journal of Alloys and Compounds 785, 320 (2019)
work page 2019
- [16]
-
[17]
V. Ladygin, P. Korotaev, A. Yanilkin, and A. Shapeev, Lattice dynamics simulation using machine learning in- teratomic potentials, Computational Materials Science 172, 109333 (2020)
work page 2020
-
[18]
J. Byggmästar, K. Nordlund, and F. Djurabekova, Modeling refractory high-entropy alloys with efficient machine-learned interatomic potentials: Defects and seg- regation, Physical Review B 104, 104101 (2021)
work page 2021
-
[19]
J. Byggmästar, K. Nordlund, and F. Djurabekova, Sim- ple machine-learned interatomic potentials for complex alloys, Physical Review Materials 6, 083801 (2022)
work page 2022
-
[20]
J. Wang, H. Kwon, H. S. Kim, and B.-J. Lee, A neural network model for high entropy alloy design, npj Com- putational Materials 9, 60 (2023)
work page 2023
-
[21]
R. Wang, X. Ma, L. Zhang, H. Wang, D. J. Srolovitz, T. Wen, and Z. Wu, Classical and machine learning in- teratomic potentials for bcc vanadium, Physical Review Materials 6, 113603 (2022)
work page 2022
-
[22]
T. Wang, J. Li, M. Wang, C. Li, Y. Su, S. Xu, and X.-G. Li, Unraveling dislocation-based strengthening in refrac- tory multi-principal element alloys, npj Computational Materials 10, 143 (2024)
work page 2024
-
[23]
Kikuchi, A theory of cooperative phenomena, Physical Review 81, 988 (1951)
R. Kikuchi, A theory of cooperative phenomena, Physical Review 81, 988 (1951)
work page 1951
-
[24]
J. M. Sanchez, F. Ducastelle, and D. Gratias, Generalized cluster description of multicomponent systems, Physica A: Statistical Mechanics and its Applications 128, 334 (1984)
work page 1984
-
[25]
D. de Fontaine, The cluster variation method and the calculation of alloy phase diagrams, in Alloy Phase Sta- bility, edited by G. M. Stocks and A. Gonis (Springer Netherlands, Dordrecht, 1989) pp. 177–203
work page 1989
- [26]
- [27]
-
[28]
S. Kadkhodaei and J. A. Muñoz, Cluster expansion of al- loy theory: A review of historical development and mod- ern innovations, JOM 73, 3326 (2021)
work page 2021
-
[29]
A. D. Kim and M. Widom, Interaction models and con- figurational entropies of binary mota and the monbtaw high entropy alloy, Physical Review Materials 7, 063803 (2023)
work page 2023
-
[30]
W. Kohn and L. J. Sham, Self-consistent equations in- cluding exchange and correlation effects, Physical Review 140, A1133 (1965)
work page 1965
-
[31]
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococ- cioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. S...
work page 2009
-
[32]
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. D. Jr, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawa- mura, H.-Y. Ko, A. Kokalj, E. Küçük...
work page 2017
-
[33]
G. Kresse and D. Joubert, From ultrasoft pseudopoten- tials to the projector augmented-wave method, Physical Review B 59, 1758 (1999)
work page 1999
-
[34]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Physical Review Letters 77, 3865 (1996)
work page 1996
-
[35]
A. Dal Corso, Pseudopotentials periodic table: From h to pu, Computational Materials Science 95, 337 (2014)
work page 2014
-
[36]
H. J. Monkhorst and J. D. Pack, Special points for brillouin-zone integrations, Physical Review B 13, 5188 (1976)
work page 1976
-
[37]
T. Xie and J. C. Grossman, Crystal graph convolutional neural networks for an accurate and interpretable predic- tion of material properties, Physical Review Letters 120, 10.1103/physrevlett.120.145301 (2018)
-
[38]
P. Veličković, G. Cucurull, A. Casanova, A. Romero, P. Liò, and Y. Bengio, Graph attention networks (2018), arXiv:1710.10903 [stat.ML]
work page internal anchor Pith review Pith/arXiv arXiv 2018
- [39]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.