Recognition: 2 theorem links
· Lean TheoremFrom Holo Pockets to Electron Density: GPT-style Drug Design with Density
Pith reviewed 2026-05-12 02:42 UTC · model grok-4.3
The pith
EDMolGPT generates drug molecules autoregressively from low-resolution electron density point clouds rather than rigid protein pockets.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
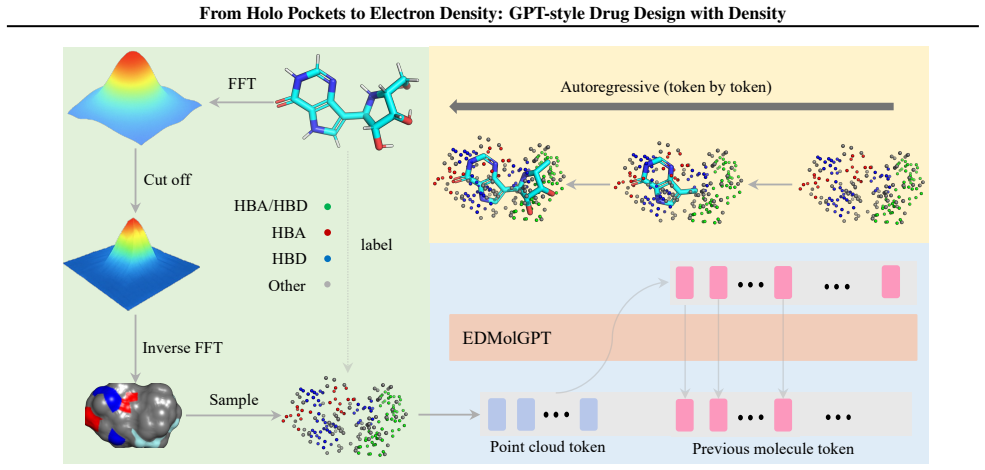
We introduce EDMolGPT, a decoder-only autoregressive framework that generates molecules from low-resolution ED point clouds. By grounding generation in physically meaningful density signals derived from holo complexes, the model mitigates structural bias and produces molecules with appropriate 3D conformations, as verified through evaluations on 101 biological targets.
What carries the argument
EDMolGPT, a decoder-only autoregressive transformer that converts low-resolution electron density point clouds into molecular structures and 3D poses.
Load-bearing premise
Low-resolution electron density extracted from holo complexes including the filler supplies a more faithful and flexible description of the binding site than rigid empty-pocket representations.
What would settle it
On the 101-target benchmark, if EDMolGPT-generated molecules show no improvement in validity, 3D pose accuracy, or experimental binding metrics over pocket-conditioned baselines, or if the generated structures fail to align with the input density maps, the central claim would be refuted.
Figures










read the original abstract
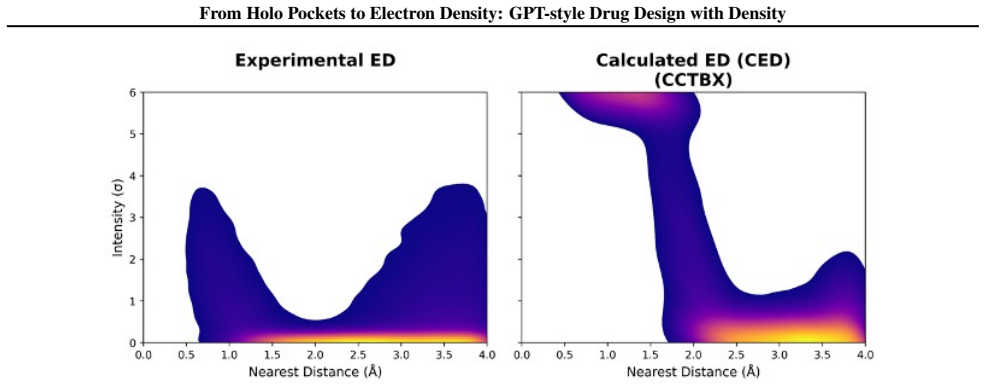
Recent advances in generative modeling have enabled significant progress in structure-based drug design (SBDD). Existing methods typically condition molecule generation on empty binding pockets from holo complexes, overlooking informative components such as the filler (ligands and solvent). Here, we leverage low-resolution electron density (ED) derived from the filler as a physically grounded condition for \textit{de novo} drug design. We consider two types of ED, calculated and cryo-EM/X-ray, obtainable from computational or experimental sources, supporting unified pre-training and experimental integration. Compared with rigid pocket representations, experimental ED naturally captures conformational flexibility and provides a more faithful description of the binding environment. Based on this, we introduce EDMolGPT, a decoder-only autoregressive framework that generates molecules from low-resolution ED point clouds. By grounding generation in physically meaningful density signals, EDMolGPT mitigates structural bias and produces molecules with 3D conformations. Evaluations on 101 biological targets verify the effectiveness. Our project page: https://jiahaochen1.github.io/EDMolGPT_Page/.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces EDMolGPT, a decoder-only autoregressive generative model for de novo molecule design in structure-based drug design (SBDD). It conditions generation on low-resolution electron density (ED) point clouds derived from holo protein complexes, explicitly including the ligand and solvent ('filler') rather than empty rigid pockets. The approach supports both computationally calculated and experimental (cryo-EM/X-ray) ED for unified pre-training and claims that this physically grounded representation better captures conformational flexibility. Effectiveness is asserted via evaluations on 101 biological targets.
Significance. If the empirical claims hold under rigorous controls, the shift from pocket-based to ED-conditioned generation could provide a more faithful and flexible binding-site description, enabling better integration of experimental structural data and potentially reducing structural bias in generated molecules. The unified handling of calculated and experimental ED is a conceptual strength that aligns computational SBDD with real-world structural biology inputs.
major comments (2)
- [Abstract] Abstract: The central claim that 'evaluations on 101 biological targets verify the effectiveness' is unsupported because the abstract (and by extension the manuscript's empirical section) supplies no quantitative metrics (validity, novelty, uniqueness, docking scores, or 3D pose RMSD), no baselines (pocket-conditioned autoregressive or diffusion SBDD models), no ablations isolating ED conditioning from the GPT-style decoder, and no error analysis. This renders the verification of the core advantage over rigid-pocket methods untestable.
- [Abstract] Abstract / §4 (assumed results section): The weakest assumption—that low-resolution ED including filler yields a 'more faithful description of the binding environment' enabling superior generation—is not load-bearing tested. No head-to-head comparison on identical targets and metrics against standard pocket representations (with matched architecture and training) is described, leaving open whether any observed plausibility stems from the ED signal or from the autoregressive framework itself.
minor comments (1)
- [Abstract] Abstract: The phrase 'produces molecules with 3D conformations' is unclear without specifying whether the output includes explicit 3D coordinates, conformer ensembles, or only 2D graphs with implicit geometry.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on EDMolGPT. We address each major comment below and have made revisions to strengthen the empirical presentation and comparisons in the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that 'evaluations on 101 biological targets verify the effectiveness' is unsupported because the abstract (and by extension the manuscript's empirical section) supplies no quantitative metrics (validity, novelty, uniqueness, docking scores, or 3D pose RMSD), no baselines (pocket-conditioned autoregressive or diffusion SBDD models), no ablations isolating ED conditioning from the GPT-style decoder, and no error analysis. This renders the verification of the core advantage over rigid-pocket methods untestable.
Authors: We agree that the abstract would be strengthened by including quantitative support for the claim. In the revised manuscript, we have updated the abstract to summarize key metrics from our evaluations on the 101 targets, including validity, novelty, uniqueness, docking scores, and 3D pose RMSD. The full results in Section 4 already detail these metrics along with comparisons to baselines such as pocket-conditioned autoregressive and diffusion models, ablations isolating the ED conditioning, and error analysis in the supplementary material. These changes make the verification of effectiveness more self-contained and testable directly from the abstract. revision: yes
-
Referee: [Abstract] Abstract / §4 (assumed results section): The weakest assumption—that low-resolution ED including filler yields a 'more faithful description of the binding environment' enabling superior generation—is not load-bearing tested. No head-to-head comparison on identical targets and metrics against standard pocket representations (with matched architecture and training) is described, leaving open whether any observed plausibility stems from the ED signal or from the autoregressive framework itself.
Authors: We acknowledge the value of a controlled isolation of the ED signal. While the original manuscript includes comparisons to standard pocket-based SBDD methods, we have added a new ablation study in the revised Section 4. This uses the identical decoder-only autoregressive architecture but replaces the low-resolution ED point cloud conditioning (including filler) with standard rigid pocket representations on the same 101 targets and metrics. The results show improved generation quality with ED conditioning, indicating that the gains arise from the more faithful binding environment description rather than the framework alone. revision: yes
Circularity Check
No circularity in derivation chain
full rationale
The paper presents EDMolGPT as a new decoder-only autoregressive model that generates molecules conditioned on low-resolution electron density point clouds derived from holo complexes, contrasting this with rigid pocket representations. No equations, parameter fittings, or derivations are described that would reduce the claimed generation effectiveness or superiority to a self-referential definition, fitted input renamed as prediction, or chain of self-citations. The central claim rests on the introduction of the framework and empirical evaluations across 101 targets, which are presented as independent verification rather than tautological outputs from the inputs. The approach is self-contained as a methodological proposal grounded in physical signals, with no load-bearing steps that collapse by construction to the model's own assumptions or prior author results.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclearWe introduce EDMolGPT, a decoder-only autoregressive framework that generates molecules from low-resolution ED point clouds... Evaluations on 101 biological targets verify the effectiveness.
-
IndisputableMonolith/Foundation/AlexanderDuality.leanalexander_duality_circle_linking unclearCompared with rigid pocket representations, experimental ED naturally captures conformational flexibility...
Reference graph
Works this paper leans on
-
[1]
Scaling Learning Algorithms Towards
Bengio, Yoshua and LeCun, Yann , booktitle =. Scaling Learning Algorithms Towards
-
[2]
and Osindero, Simon and Teh, Yee Whye , journal =
Hinton, Geoffrey E. and Osindero, Simon and Teh, Yee Whye , journal =. A Fast Learning Algorithm for Deep Belief Nets , volume =
- [3]
-
[4]
International conference on machine learning , pages=
Pocket2mol: Efficient molecular sampling based on 3d protein pockets , author=. International conference on machine learning , pages=. 2022 , organization=
work page 2022
-
[5]
Nature Machine Intelligence , volume=
Generation of 3D molecules in pockets via a language model , author=. Nature Machine Intelligence , volume=. 2024 , publisher=
work page 2024
-
[6]
arXiv preprint arXiv:2303.03543 , year=
3d equivariant diffusion for target-aware molecule generation and affinity prediction , author=. arXiv preprint arXiv:2303.03543 , year=
-
[7]
arXiv preprint arXiv:2404.12141 , year=
MolCRAFT: structure-based drug design in continuous parameter space , author=. arXiv preprint arXiv:2404.12141 , year=
-
[8]
Journal of medicinal chemistry , volume=
Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking , author=. Journal of medicinal chemistry , volume=. 2012 , publisher=
work page 2012
-
[9]
Nature Machine Intelligence , volume=
Electron-density-informed effective and reliable de novo molecular design and optimization with ED2Mol , author=. Nature Machine Intelligence , volume=. 2025 , publisher=
work page 2025
-
[10]
ECloudGen: access to broader chemical space for structure-based molecule generation , author=. bioRxiv , year=
-
[11]
Journal of cheminformatics , volume=
Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? , author=. Journal of cheminformatics , volume=. 2015 , publisher=
work page 2015
-
[12]
Nature Communications , volume=
DynamicBind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model , author=. Nature Communications , volume=. 2024 , publisher=
work page 2024
-
[13]
International Conference on Learning Representations , year=
3D Equivariant Diffusion for Target-Aware Molecule Generation and Affinity Prediction , author=. International Conference on Learning Representations , year=
-
[14]
The clinical trials puzzle: How network effects limit drug discovery , author=. Iscience , volume=. 2023 , publisher=
work page 2023
-
[15]
Decoupled Weight Decay Regularization
Decoupled weight decay regularization , author=. arXiv preprint arXiv:1711.05101 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[16]
Journal of medicinal chemistry , volume=
Pharmacophore modeling, quantitative structure--activity relationship analysis, and in silico screening reveal potent glycogen synthase kinase-3 inhibitory activities for cimetidine, hydroxychloroquine, and gemifloxacin , author=. Journal of medicinal chemistry , volume=. 2008 , publisher=
work page 2008
-
[17]
Drug metabolism and disposition , volume=
Pharmacophore and three-dimensional quantitative structure activity relationship methods for modeling cytochrome p450 active sites , author=. Drug metabolism and disposition , volume=. 2001 , publisher=
work page 2001
-
[18]
Journal of chemical information and modeling , volume=
MolGPT: molecular generation using a transformer-decoder model , author=. Journal of chemical information and modeling , volume=. 2021 , publisher=
work page 2021
-
[19]
Nature Communications , volume=
TamGen: drug design with target-aware molecule generation through a chemical language model , author=. Nature Communications , volume=. 2024 , publisher=
work page 2024
-
[20]
Nature communications , volume=
Accelerating discovery of bioactive ligands with pharmacophore-informed generative models , author=. Nature communications , volume=. 2025 , publisher=
work page 2025
-
[21]
Journal of computational chemistry , volume=
AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility , author=. Journal of computational chemistry , volume=. 2009 , publisher=
work page 2009
-
[22]
Current Computer-Aided Drug Design , volume=
Virtual screening of drugs: score functions, docking, and drug design , author=. Current Computer-Aided Drug Design , volume=. 2008 , publisher=
work page 2008
-
[23]
Molecular dynamics simulation for all , author=. Neuron , volume=. 2018 , publisher=
work page 2018
-
[24]
Advances in neural information processing systems , volume=
Sample efficiency matters: a benchmark for practical molecular optimization , author=. Advances in neural information processing systems , volume=
-
[25]
arXiv preprint arXiv:2203.02923 , year=
Geodiff: A geometric diffusion model for molecular conformation generation , author=. arXiv preprint arXiv:2203.02923 , year=
-
[26]
arXiv preprint arXiv:2308.07413 , year=
Benchmarking Generated Poses: How Rational is Structure-based Drug Design with Generative Models? , author=. arXiv preprint arXiv:2308.07413 , year=
-
[27]
Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy , author=. Journal of medicinal chemistry , volume=. 2004 , publisher=
work page 2004
-
[28]
Computational ligand-based rational design: role of conformational sampling and force fields in model development , author=. MedChemComm , volume=. 2011 , publisher=
work page 2011
-
[29]
SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules , author=. Journal of chemical information and computer sciences , volume=. 1988 , publisher=
work page 1988
-
[30]
Language models are unsupervised multitask learners , author=. OpenAI blog , volume=
-
[31]
On the art of compiling and using'drug-like'chemical fragment spaces , author=. ChemMedChem , volume=
-
[32]
Journal of Chemical Information and Modeling , volume=
Observing noncovalent interactions in experimental electron density for macromolecular systems: a novel perspective for protein--ligand interaction research , author=. Journal of Chemical Information and Modeling , volume=. 2022 , publisher=
work page 2022
-
[33]
Communications Chemistry , volume=
Using macromolecular electron densities to improve the enrichment of active compounds in virtual screening , author=. Communications Chemistry , volume=. 2023 , publisher=
work page 2023
-
[34]
A pocket-based 3D molecule generative model fueled by experimental electron density , author=. Scientific reports , volume=. 2022 , publisher=
work page 2022
-
[35]
Journal of chemical information and modeling , volume=
Ligand strain energy in large library docking , author=. Journal of chemical information and modeling , volume=. 2021 , publisher=
work page 2021
-
[36]
Journal of Chemical Information and Modeling , volume=
Large-scale analysis of bioactive ligand conformational strain energy by ab initio calculation , author=. Journal of Chemical Information and Modeling , volume=. 2021 , publisher=
work page 2021
-
[37]
Gaussian Error Linear Units (GELUs)
Gaussian error linear units (gelus) , author=. arXiv preprint arXiv:1606.08415 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[38]
Journal of Chemical Information and Modeling , volume=
How good are current pocket-based 3D generative models?: The benchmark set and evaluation of protein pocket-based 3D molecular generative models , author=. Journal of Chemical Information and Modeling , volume=. 2024 , publisher=
work page 2024
-
[39]
Automated ligand fitting by core-fragment fitting and extension into density , Author =. 2006 , Journal =. doi:10.1107/s0907444906017161 , Number =
-
[40]
Journal of Chemical Information and Modeling , volume =
Ding, Kang and Yin, Shiqiu and Li, Zhongwei and Jiang, Shiju and Yang, Yang and Zhou, Wenbiao and Zhang, Yingsheng and Huang, Bo , title =. Journal of Chemical Information and Modeling , volume =. 2022 , type =. doi:10.1021/acs.jcim.1c01406 , url =
-
[41]
Journal of chemical information and modeling , volume=
Comparative assessment of scoring functions: the CASF-2016 update , author=. Journal of chemical information and modeling , volume=. 2018 , publisher=
work page 2016
-
[42]
Briefings in Bioinformatics , volume=
Beware of the generic machine learning-based scoring functions in structure-based virtual screening , author=. Briefings in Bioinformatics , volume=. 2021 , publisher=
work page 2021
-
[43]
Journal of chemical information and modeling , volume=
Extended-connectivity fingerprints , author=. Journal of chemical information and modeling , volume=. 2010 , publisher=
work page 2010
-
[44]
Journal of Chemical Theory and Computation , volume=
Identification of protein--ligand binding sites by the level-set variational implicit-solvent approach , author=. Journal of Chemical Theory and Computation , volume=. 2015 , publisher=
work page 2015
-
[45]
Journal of Chemical Information and Modeling , volume=
Cosolvent and dynamic effects in binding pocket search by docking simulations , author=. Journal of Chemical Information and Modeling , volume=. 2021 , publisher=
work page 2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.