Recognition: no theorem link
Accelerating Locality-Driven Integration in Quantum Chemistry with Block-Structured Matrix Multiplication
Pith reviewed 2026-05-14 21:31 UTC · model grok-4.3
The pith
KerneLDI reorganizes matrix data into block-filtered form to accelerate locality-driven integration in quantum chemistry by up to 10 times on GPUs while keeping numerical accuracy intact.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
KerneLDI reorganizes operand matrices into a unified block-filtered representation that retains only spatially relevant blocks and executes the resulting contractions with customized dense block multipliers that adapt proven dense-matmul optimizations to retained block pairs, thereby delivering up to 10 times speedup for exchange-correlation evaluation over a dense GPU baseline while preserving numerical accuracy.
What carries the argument
Unified block-filtered matrix representation together with customized dense block multipliers applied only to retained block pairs.
If this is right
- Numerical accuracy is preserved for exchange-correlation integration across tested molecular systems.
- Up to 10 times speedup is observed for exchange-correlation evaluation relative to a dense GPU baseline.
- Performance scales favorably as molecular system size grows and when multiple GPUs are used.
- End-to-end self-consistent field calculations run faster under the new framework.
- Ab initio molecular dynamics achieves nearly 6 times higher throughput.
Where Pith is reading between the lines
- The same block-filtered layout could be applied to other integral-screening or quadrature steps that exhibit spatial locality.
- Further gains may appear in even larger systems where the fraction of discarded blocks increases.
- The co-design pattern of screening logic plus dense-block kernels might transfer to related sparse-dense hybrid computations outside quantum chemistry.
- Porting the approach to other GPU architectures would test whether the reported speedups depend on specific hardware features.
Load-bearing premise
Reorganizing matrices into a unified block-filtered representation retains every spatially relevant contribution without numerical error or extra problem-specific tuning.
What would settle it
Compute exchange-correlation energies or forces for a large molecular system with both KerneLDI and an unmodified dense GPU baseline; any deviation larger than floating-point tolerance or any speedup below the claimed factor on that system would falsify the central claim.
Figures






read the original abstract
Locality-driven integration is a pervasive computational pattern in quantum chemistry, arising whenever spatially localized basis functions interact through numerical quadrature or integral screening. The dominant matrix multiplications in these tasks exhibit dynamic, structured sparsity driven by spatial locality, posing significant challenges for both dense batched kernels and generic sparse formats on GPUs. We present KerneLDI, a GPU-oriented framework that addresses this regime by co-designing data layout, screening logic, and matrix-computation operators to realize block-structured matrix multiplication for locality-driven integration. KerneLDI reorganizes operand matrices into a unified block-filtered representation that retains only spatially relevant blocks, and executes the resulting contractions with customized dense block multipliers that adapt proven dense-matmul optimizations to retained block pairs. We develop and evaluate KerneLDI on exchange--correlation (EXC) integration in Kohn--Sham density functional theory, a representative and computationally critical instance of this pattern. Across diverse molecular systems, KerneLDI preserves numerical accuracy while delivering up to 10$\times$ speedup for EXC evaluation over a dense GPU baseline, scales favorably with increasing system size and multi-GPU parallelism, accelerates end-to-end self-consistent field calculations, and yields nearly 6$\times$ throughput improvement for ab initio molecular dynamics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces KerneLDI, a GPU-oriented framework that co-designs data layout, screening logic, and matrix operators to perform block-structured matrix multiplication for locality-driven integration tasks in quantum chemistry. Focused on exchange-correlation (EXC) integration within Kohn-Sham DFT, the approach reorganizes operand matrices into a unified block-filtered representation retaining only spatially relevant blocks and executes contractions via customized dense block multipliers. The authors claim that numerical accuracy is preserved across tested molecular systems while delivering up to 10× speedup for EXC evaluation versus a dense GPU baseline, favorable scaling with system size and multi-GPU parallelism, acceleration of end-to-end SCF calculations, and nearly 6× throughput gains for ab initio molecular dynamics.
Significance. If the performance and accuracy claims are substantiated with detailed verification, this work addresses a pervasive computational pattern in quantum chemistry by mapping locality-driven sparsity onto efficient GPU kernels without generic sparse formats. It could meaningfully improve throughput for DFT-based simulations and molecular dynamics on modern hardware, particularly as system sizes grow, and the co-design strategy may generalize to other quadrature or integral-screening workloads.
major comments (2)
- [Abstract and §4] Abstract and §4 (Evaluation): the central claim that numerical accuracy is preserved lacks reported error bars, quantitative thresholds for the screening logic, or explicit comparison against established sparse GPU libraries such as cuSPARSE; without these, the 10× speedup cannot be fully assessed for hidden costs or generality.
- [§3] §3 (Methods): the assumption that the block-filtered representation retains all spatially relevant contributions exactly is load-bearing for the accuracy claim, yet no formal argument or exhaustive edge-case testing (e.g., systems near locality breakdown) is provided to confirm absence of truncation errors beyond the tested molecules.
minor comments (2)
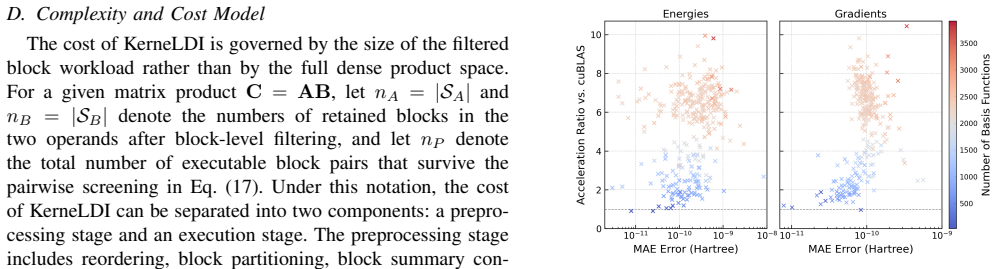
- [§4.2] Figure captions and §4.2 should explicitly state the molecular systems, basis sets, and functional used in the timing and accuracy benchmarks for reproducibility.
- [§3] Notation for block indices and screening parameters could be unified across equations and pseudocode to avoid minor ambiguity in the operator definitions.
Simulated Author's Rebuttal
We thank the referee for the constructive review and the recommendation for minor revision. We appreciate the recognition of the potential impact of KerneLDI on locality-driven integration tasks in quantum chemistry. We address each major comment below and have incorporated revisions to strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract and §4] Abstract and §4 (Evaluation): the central claim that numerical accuracy is preserved lacks reported error bars, quantitative thresholds for the screening logic, or explicit comparison against established sparse GPU libraries such as cuSPARSE; without these, the 10× speedup cannot be fully assessed for hidden costs or generality.
Authors: We agree that error bars, explicit screening thresholds, and a comparison to cuSPARSE would improve the assessment of accuracy and performance claims. In the revised manuscript we have added error bars to all accuracy metrics in §4, stated the quantitative screening thresholds used to construct the block-filtered representation, and included direct benchmarks against cuSPARSE. These additions confirm that the observed speedups incur no hidden accuracy penalties relative to the dense baseline or generic sparse libraries for the targeted structured-sparsity regime. revision: yes
-
Referee: [§3] §3 (Methods): the assumption that the block-filtered representation retains all spatially relevant contributions exactly is load-bearing for the accuracy claim, yet no formal argument or exhaustive edge-case testing (e.g., systems near locality breakdown) is provided to confirm absence of truncation errors beyond the tested molecules.
Authors: The block-filtered representation retains blocks according to a spatial-overlap criterion that follows established locality principles in quantum chemistry; by construction it excludes only blocks whose contribution falls below the chosen threshold. While a fully general formal proof is difficult because locality is itself an approximation, we have expanded §3 with a detailed derivation of the retention criterion and its relation to standard integral-screening bounds. We have also added supplementary experiments on systems near the locality limit (highly delocalized and extended molecules) and report that truncation errors remain negligible within the tested regimes, consistent with the original accuracy results. revision: yes
Circularity Check
No significant circularity; implementation framework is self-contained
full rationale
The paper describes an engineering co-design of data layout, screening, and dense block matrix kernels for locality-driven integration in quantum chemistry (EXC evaluation in DFT). No mathematical derivation chain exists that reduces predictions or results to fitted parameters, self-definitions, or self-citation load-bearing steps. Claims rest on empirical benchmarks showing preserved numerical accuracy and measured speedups across systems, with the block-filtered representation and operators presented as direct mappings of existing spatial locality patterns onto GPU primitives. The central contribution is algorithmic implementation rather than a first-principles result derived from its own outputs.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
J. Nickolls and W. J. Dally, “GPU computing,”Proceedings of the IEEE, vol. 98, no. 8, pp. 1479–1492, 2010
work page 2010
-
[2]
Benchmarking GPUs to tune dense linear algebra,
V . V olkov and J. W. Demmel, “Benchmarking GPUs to tune dense linear algebra,” inSC ’08: Proceedings of the 2008 ACM/IEEE Conference on Supercomputing. IEEE, 2008, pp. 1–11. [Online]. Available: https://doi.org/10.1109/SC.2008.5214359
-
[3]
cublas: The nvidia cuda basic linear algebra subroutine library,
“cublas: The nvidia cuda basic linear algebra subroutine library,” https: //docs.nvidia.com/cuda/cublas/, 2024, accessed: 2024-11-11
work page 2024
-
[4]
nvidia.com/cuda/cuda-c-programming-guide/
NVIDIA Corporation,CUDA C Programming Guide, 2019, https://docs. nvidia.com/cuda/cuda-c-programming-guide/
work page 2019
-
[5]
Implementing sparse matrix-vector multiplication on throughput-oriented processors,
N. Bell and M. Garland, “Implementing sparse matrix-vector multiplication on throughput-oriented processors,” inProceedings of the Conference on High Performance Computing Networking, Storage and Analysis (SC ’09), 2009, pp. 1–11. [Online]. Available: https://doi.org/10.1145/1654059.1654078
-
[6]
M. Naumov, L. Chien, P. Vandermersch, and U. Kapasi, “Cusparse library,” inGPU Technology Conference, vol. 12, 2010
work page 2010
-
[7]
Sparse matrix-vector multiplication on GPGPUs,
S. Filippone, V . Cardellini, D. Barbieri, and A. Luque, “Sparse matrix-vector multiplication on GPGPUs,”ACM Transactions on Mathematical Software, vol. 43, no. 4, pp. 1–49, 2017. [Online]. Available: https://doi.org/10.1145/3017994
-
[8]
Parallel sparse matrix-matrix multiplication and indexing: Implementation and experiments,
A. Buluc ¸ and J. R. Gilbert, “Parallel sparse matrix-matrix multiplication and indexing: Implementation and experiments,”SIAM Journal on Scientific Computing, vol. 34, no. 4, pp. C170–C191, 2012. [Online]. Available: https://doi.org/10.1137/110848244
-
[9]
Design principles for sparse matrix multiplication on the GPU,
C. Yang, A. Buluc ¸, and J. D. Owens, “Design principles for sparse matrix multiplication on the GPU,” inEuro-Par 2018: Parallel Processing. Springer, 2018, pp. 672–687. [Online]. Available: https://doi.org/10.1007/978-3-319-96983-1 48
-
[10]
The University of Florida sparse matrix collection,
T. A. Davis and Y . Hu, “The University of Florida sparse matrix collection,”ACM Transactions on Mathematical Software, vol. 38, no. 1, pp. 1–25, 2011. [Online]. Available: https://doi.org/10.1145/ 2049662.2049663
-
[11]
Fast sparse matrix-vector multiplication by exploiting variable block structure,
R. W. Vuduc and H.-J. Moon, “Fast sparse matrix-vector multiplication by exploiting variable block structure,” inHigh Performance Computing and Communications: First International Conference, HPCC 2005, Sorrento, Italy, September 21-23, 2005. Proceedings 1. Springer, 2005, pp. 807–816. [Online]. Available: https://doi.org/10.1007/11557654 91
-
[12]
Optimization of block sparse matrix- vector multiplication on shared-memory parallel architectures,
R. Eberhardt and M. Hoemmen, “Optimization of block sparse matrix- vector multiplication on shared-memory parallel architectures,” in2016 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW). IEEE, 2016, pp. 663–672. [Online]. Available: https://doi.org/10.1109/IPDPSW.2016.42
-
[13]
Integral approximations for lcao-scf calculations,
O. Vahtras, J. Alml ¨of, and M. W. Feyereisen, “Integral approximations for lcao-scf calculations,”Chemical Physics Letters, vol. 213, no. 5–6, pp. 514–518, 1993. [Online]. Available: https://doi.org/10.1016/ 0009-2614(93)89151-7
work page 1993
-
[14]
J. Kussmann, H. Laqua, and C. Ochsenfeld, “Highly efficient resolution-of-identity density functional theory calculations on central and graphics processing units,”Journal of Chemical Theory and Computation, vol. 17, no. 3, pp. 1512–1521, 2021. [Online]. Available: https://doi.org/10.1021/acs.jctc.0c01252
-
[15]
Highly efficient, linear-scaling seminumerical exact-exchange method for graphic processing units,
H. Laqua, T. H. Thompson, J. Kussmann, and C. Ochsenfeld, “Highly efficient, linear-scaling seminumerical exact-exchange method for graphic processing units,”Journal of Chemical Theory and Computation, vol. 16, no. 3, pp. 1456–1468, 2020. [Online]. Available: https://doi.org/10.1021/acs.jctc.9b00860
-
[16]
M. Manathunga, Y . Miao, D. Mu, A. W. G”otz, and K. M. Merz, Jr., “Parallel implementation of density functional theory methods in the quantum interaction computational kernel program,”Journal of Chemical Theory and Computation, vol. 16, no. 7, pp. 4315–4326,
-
[17]
Available: https://doi.org/10.1021/acs.jctc.0c00290
[Online]. Available: https://doi.org/10.1021/acs.jctc.0c00290
-
[18]
D. B. Williams-Young, A. Bagusetty, W. A. de Jong, D. Doerfler, H. J. J. van Dam, ´A. V ´azquez-Mayagoitia, T. L. Windus, and C. Yang, “Achieving performance portability in gaussian basis set density functional theory on accelerator based architectures in nwchemex,” Parallel Computing, vol. 108, p. 102829, 2021. [Online]. Available: https://doi.org/10.101...
-
[19]
Enhancing gpu-acceleration in the python-based simulations of chemistry frameworks,
X. Wu, Q. Sun, Z. Pu, T. Zheng, W. Ma, W. Yan, Y . Xia, Z. Wu, M. Huo, X. Liet al., “Enhancing gpu-acceleration in the python-based simulations of chemistry frameworks,”Wiley Interdisciplinary Reviews: Computational Molecular Science, vol. 15, no. 2, p. e70008, 2025. [Online]. Available: https://doi.org/10.1002/wcms.70008
-
[20]
Efficient algorithms for gpu accelerated evaluation of the dft exchange-correlation functional,
R. Stocks and G. M. Barca, “Efficient algorithms for gpu accelerated evaluation of the dft exchange-correlation functional,”Journal of Chemical Theory and Computation, vol. 21, no. 20, pp. 10 263–10 280,
-
[21]
Available: https://doi.org/10.1021/acs.jctc.5c01229
[Online]. Available: https://doi.org/10.1021/acs.jctc.5c01229
-
[22]
A multicenter numerical integration scheme for polyatomic molecules,
A. D. Becke, “A multicenter numerical integration scheme for polyatomic molecules,”The Journal of Chemical Physics, vol. 88, no. 4, pp. 2547–2553, 1988. [Online]. Available: https://doi.org/10. 1063/1.454033
work page 1988
-
[23]
Efficient molecular numerical integration schemes,
O. Treutler and R. Ahlrichs, “Efficient molecular numerical integration schemes,”The Journal of Chemical Physics, vol. 102, no. 1, pp. 346–354, 1995. [Online]. Available: https://doi.org/10.1063/1.469408
-
[24]
A standard grid for density functional calculations,
P. M. W. Gill, B. G. Johnson, and J. A. Pople, “A standard grid for density functional calculations,”Chemical Physics Letters, vol. 209, no. 5–6, pp. 506–512, 1993. [Online]. Available: https: //doi.org/10.1016/0009-2614(93)80125-9
-
[25]
D. Marx and J. Hutter,Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods. Cambridge University Press, 2009
work page 2009
-
[26]
G. Kresse and J. Furthm ¨uller, “Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set,”Physical Review B, vol. 54, no. 16, pp. 11 169–11 186, 1996. [Online]. Available: https://doi.org/10.1103/PhysRevB.54.11169
-
[27]
Cp2k: atomistic simulations of condensed matter systems,
J. Hutter, M. Iannuzzi, F. Schiffmann, and J. VandeV ondele, “Cp2k: atomistic simulations of condensed matter systems,”WIREs Computational Molecular Science, vol. 4, no. 1, pp. 15–25, 2014. [Online]. Available: https://doi.org/10.1002/wcms.1159
-
[28]
Acceleration without disruption: Dft software as a service,
F. Ju, X. Wei, L. Huang, A. J. Jenkins, L. Xia, J. Zhang, J. Zhu, H. Yang, B. Shao, P. Dai, D. B. Williams-Young, A. Mayya, Z. Hooshmand, A. Efimovskaya, N. A. Baker, M. Troyer, and H. Liu, “Acceleration without disruption: Dft software as a service,”Journal of Chemical Theory and Computation, 2024. [Online]. Available: https://doi.org/10.1021/acs.jctc.4c00940
-
[29]
Y . Zhao and D. G. Truhlar, “The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: 11 two new functionals and systematic testing of four m06- class functionals and 12 other functionals,”Theoretical chemistry accounts, vol. 120, no. 1, pp. 215–241, 20...
-
[30]
F. Weigend and R. Ahlrichs, “Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy,”Physical Chemistry Chemical Physics, vol. 7, no. 18, pp. 3297–3305, 2005. [Online]. Available: https://doi.org/10.1039/b508541a
-
[31]
Extending sparse tensor accelerators to support multiple compression formats,
E. Qin, G. Jeong, W. Won, S.-C. Kao, H. Kwon, S. Srinivasan, D. Das, G. E. Moon, S. Rajamanickam, and T. Krishna, “Extending sparse tensor accelerators to support multiple compression formats,” in2021 IEEE International Parallel and Distributed Processing Symposium (IPDPS). IEEE, 2021, pp. 1014–1024. [Online]. Available: https://doi.org/10.1109/IPDPS49936...
-
[32]
Sparse approximate matrix- matrix multiplication for density matrix purification with error control,
A. G. Artemov and E. H. Rubensson, “Sparse approximate matrix- matrix multiplication for density matrix purification with error control,” Journal of Computational Physics, vol. 438, p. 110354, 2021. [Online]. Available: https://doi.org/10.1016/j.jcp.2021.110354
-
[33]
Nvidia a100 tensor core gpu: Performance and innovation,
J. Choquette, W. Gandhi, O. Giroux, N. Stam, and R. Krashinsky, “Nvidia a100 tensor core gpu: Performance and innovation,”IEEE Micro, vol. 41, no. 2, pp. 29–35, 2021. [Online]. Available: https://doi.org/10.1109/MM.2021.3061394
-
[34]
Recent developments in the general atomic and molecular electronic structure system,
G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunber, E. Guidezet al., “Recent developments in the general atomic and molecular electronic structure system,”The Journal of Chemical Physics, vol. 152, no. 15, p. 154102, 2020. [Online]. Available: https://doi.org/10.1063/5.0005188
-
[35]
Nvidia tensor core programmability, performance & precision,
S. Markidis, S. W. Der Chien, E. Laure, I. B. Peng, and J. S. Vetter, “Nvidia tensor core programmability, performance & precision,” in 2018 IEEE international parallel and distributed processing symposium workshops (IPDPSW). IEEE, 2018, pp. 522–531. [Online]. Available: https://doi.org/10.1109/IPDPSW.2018.00091
-
[36]
A. Haidar, S. Tomov, J. Dongarra, and N. J. Higham, “Harnessing GPU tensor cores for fast FP16 arithmetic to speed up mixed- precision iterative refinement solvers,”Proceedings of the International Conference for High Performance Computing, Networking, Storage, and Analysis (SC ’18), pp. 1–11, 2018. [Online]. Available: https://doi.org/10.1109/SC.2018.00050
-
[37]
Ginkgo: A modern linear operator algebra framework for high performance computing,
H. Anzt, T. Cojean, G. Flegar, F. G ¨obel, T. Gr ¨utzmacher, P. Nayak, T. Ribizel, Y . M. Tsai, and E. S. Quintana-Ort ´ı, “Ginkgo: A modern linear operator algebra framework for high performance computing,” ACM Transactions on Mathematical Software, vol. 48, no. 1, pp. 1–33,
-
[38]
Available: https://doi.org/10.1145/3480935
[Online]. Available: https://doi.org/10.1145/3480935
-
[39]
Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born,
A. W. G ¨otz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand, and R. C. Walker, “Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born,”Journal of Chemical Theory and Computation, vol. 8, no. 5, pp. 1542–1555, 2012. [Online]. Available: https://doi.org/10.1021/ct200909j
-
[40]
Whippletree: task-based scheduling of dynamic workloads on the GPU,
M. Steinberger, M. Kenzel, P. Boechat, B. Kerber, M. Dokter, and D. Schmalstieg, “Whippletree: task-based scheduling of dynamic workloads on the GPU,” inACM Transactions on Graphics (TOG), vol. 33, no. 6, 2014, pp. 1–11. [Online]. Available: https://doi.org/10.1145/2661229.2661250
-
[41]
Self-consistent equations including exchange and correlation effects,
W. Kohn and L. J. Sham, “Self-consistent equations including exchange and correlation effects,”Physical Review, vol. 140, no. 4A, pp. A1133–A1138, 1965. [Online]. Available: https://doi.org/10.1103/ PhysRev.140.A1133
work page 1965
-
[42]
V . I. Lebedev, “Quadratures on a sphere,”USSR Computational Mathematics and Mathematical Physics, vol. 16, no. 2, pp. 10–24, 1976. [Online]. Available: https://doi.org/10.1016/0041-5553(76)90100-2
-
[43]
Quantum chemistry on graphical processing units. 2. direct self-consistent-field implementation,
I. S. Ufimtsev and T. J. Martinez, “Quantum chemistry on graphical processing units. 2. direct self-consistent-field implementation,”Journal of Chemical Theory and Computation, vol. 5, no. 10, pp. 2619–2628,
-
[44]
Available: https://doi.org/10.1021/ct800526s
[Online]. Available: https://doi.org/10.1021/ct800526s
-
[45]
Accelerating density functional calculations with graphics processing unit,
K. Yasuda, “Accelerating density functional calculations with graphics processing unit,”Journal of Chemical Theory and Computation, vol. 4, no. 8, pp. 1230–1236, 2008. [Online]. Available: https: //doi.org/10.1021/ct8001046
-
[46]
Transition-potential coupled cluster II: optimisation of the core orbital occupation number
J. L. G ´alvez Vallejo, G. M. J. Barca, and M. S. Gordon, “High-performance gpu-accelerated evaluation of electron repulsion integrals,”Molecular Physics, 2022. [Online]. Available: https: //doi.org/10.1080/00268976.2022.2112987
-
[47]
M. Zhou and J. Wu, “A gpu implementation of classical density functional theory for rapid prediction of gas adsorption in nanoporous materials,”The Journal of Chemical Physics, vol. 153, no. 7, 2020. [Online]. Available: https://doi.org/10.1063/5.0020797 12 APPENDIXA SUPPLEMENTARYTECHNICALDETAILS A. DFT Primer for the HPC Audience This appendix provides a...
-
[48]
Grid-Point Morton Ordering:Grid points are reordered using a Z-order (Morton) space-filling curve to concentrate spatially nearby points into contiguous memory ranges. Each grid point with Cartesian coordinates(x, y, z)is first scaled to an integer lattice by multiplying by a resolution factor (128 in our implementation) and truncating to integer values. ...
-
[49]
Overlap-Signature Construction and Basis-Function Clustering:Basis functions are reordered by clustering their overlap signatures, as outlined in Section III-B. The overlap matrixS(Eq. (8)) is already available from the Kohn– Sham setup, so no additional integral evaluation is required. For each basis functioni, the overlap signatures i = (Si1, Si2, . . ....
-
[50]
to density-fitting approaches for Coulomb and exchange terms [42]. Parallel GPU implementations have also been demonstrated for classical density functional theory [43]. For EXC integration specifically, Williams-Young et al. demon- strated efficient GPU execution within the GauXC framework by grouping grid batches into dense sub-matrices and dis- patchin...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.