Recognition: 2 theorem links
· Lean TheoremMolecular Mechanisms of Urea Interactions with Bovine Serum Albumin in an Acid-Expanded Conformation (pH 3.7)
Pith reviewed 2026-05-12 05:00 UTC · model grok-4.3
The pith
Urea induces a concentration-dependent dehydration then rehydration of the protein surface in acid-expanded bovine serum albumin while largely preserving secondary structure.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
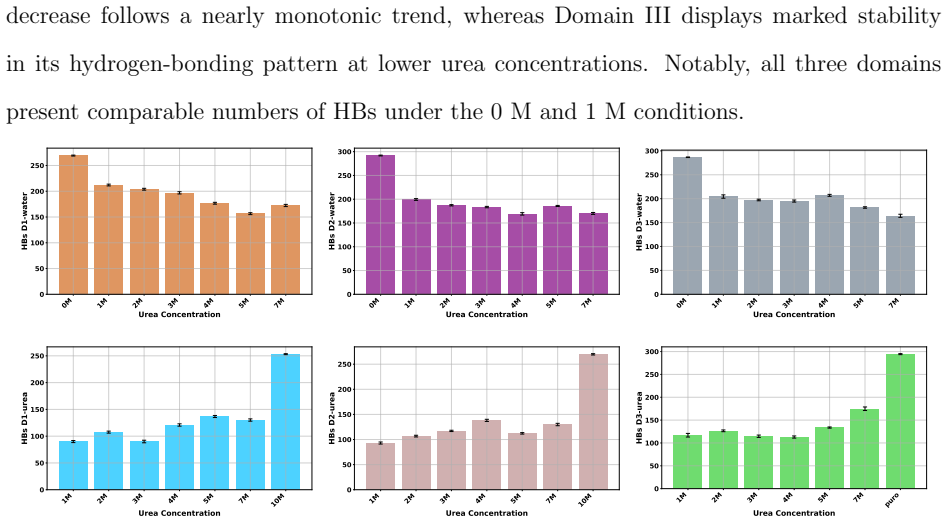
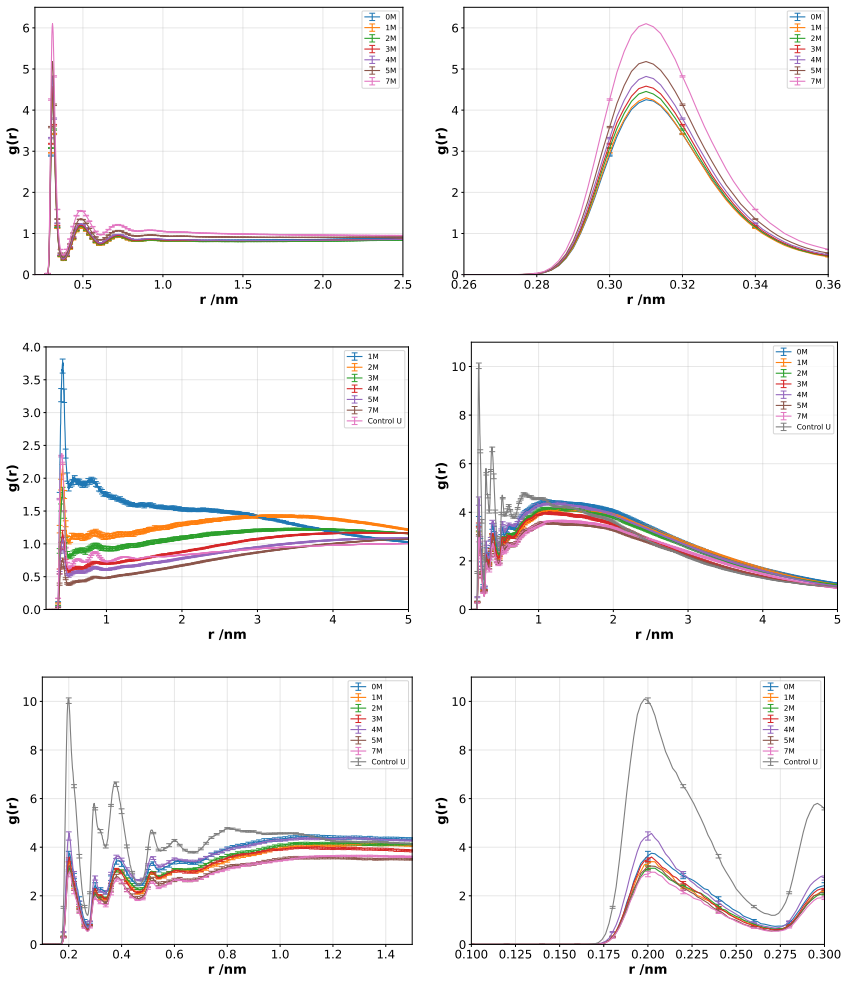
Molecular dynamics simulations of the F isoform of bovine serum albumin at pH 3.7 show that urea produces a concentration-dependent shift in the protein hydration shell: low urea concentrations reduce protein-water hydrogen bonds while increasing direct protein-urea contacts, consistent with competitive solvation; at higher concentrations urea self-association limits further protein-urea contacts and permits partial rehydration. Secondary structure elements remain largely intact, yet local and tertiary features, especially in Domain III, gain solvent exposure and conformational flexibility. These observations support a dynamic compensation mechanism in which urea partially substitutes for水水水
What carries the argument
The concentration-dependent dehydration/rehydration cycle within the protein hydration shell, driven by the balance between protein-urea contacts and urea-urea self-association.
If this is right
- Low urea concentrations act mainly through competitive displacement of water from the protein surface.
- High urea concentrations self-limit their direct interaction with the protein because urea molecules prefer to cluster with one another.
- Secondary-structure hydrogen bonds inside the protein are more resistant to urea than surface solvation contacts.
- Domain III experiences the largest increase in conformational sampling and solvent access under denaturing conditions.
- The hydrogen-bonding network around the protein can be maintained even while the composition of the solvation shell changes.
Where Pith is reading between the lines
- The same solvation-compensation pattern may operate in other multi-domain serum proteins when exposed to urea near their isoelectric points.
- Solvent engineering that modulates urea clustering could be used to tune surface hydration of proteins without triggering full unfolding.
- The observed rehydration at high urea suggests a natural upper limit to urea's denaturing power that could be tested by varying pH or adding co-solutes.
- If the mechanism holds, it predicts that mutations increasing Domain III rigidity would reduce the flexibility increase seen in the simulations.
Load-bearing premise
The chosen molecular dynamics force fields and simulation protocols correctly reproduce the real balance of urea-protein, urea-urea, and protein-water interactions for the acid-expanded F form of bovine serum albumin.
What would settle it
Direct experimental measurement, such as neutron scattering or infrared spectroscopy, of the number of protein-water versus protein-urea hydrogen bonds at several urea concentrations that fails to match the simulated trends at pH 3.7.
Figures












read the original abstract
Understanding the molecular mechanism by which denaturants modulate protein structure remains a central challenge in protein biophysics. In this work, molecular dynamics simulations were employed to investigate the effects of urea on the structural stability of bovine serum albumin, its F isoform at pH 3.7, over a broad range of urea concentrations (0 M to a fully urea/solvated system). The results reveal that urea induces a concentration/dependent dehydration/rehydration mechanism within the protein hydration shell. At low urea concentrations, a marked reduction in protein/water hydrogen bonds is observed, accompanied by a corresponding increase in protein/urea interactions, consistent with a competitive solvation process. At higher concentrations, urea/urea self-association becomes significant, limiting direct protein/urea interactions and promoting partial rehydration of the protein surface. Despite these solvent rearrangements, the secondary structure of BSA remains largely preserved, whereas local and tertiary structural features, particularly in Domain III, exhibit increased solvent exposure and conformational flexibility. These findings support a dynamic compensation mechanism in which urea partially replaces water in the solvation shell without fully disrupting the hydrogen-bonding network. Overall, this study provides molecular-level insight into the interplay between preferential interactions, solvation dynamics, and protein stability under denaturing conditions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports molecular dynamics simulations of the F isoform of bovine serum albumin at pH 3.7 in the presence of urea over a wide concentration range (0 M to fully urea-solvated). It claims that urea triggers a concentration-dependent dehydration/rehydration cycle in the protein hydration shell: at low urea, protein-water hydrogen bonds decrease while protein-urea contacts increase; at high urea, urea self-association reduces direct protein-urea interactions and allows partial rehydration. Secondary structure remains largely intact, but Domain III exhibits increased solvent exposure and flexibility, supporting a dynamic compensation mechanism in which urea partially substitutes for water without fully disrupting the hydrogen-bond network.
Significance. If the simulations accurately capture the relevant interactions and conformational ensemble, the work supplies atomistic detail on how urea modulates solvation and local structure in an acid-expanded protein without global unfolding. This could help discriminate between competing models of denaturant action (preferential binding vs. indirect effects) and is relevant to understanding protein stability under non-native conditions.
major comments (3)
- [Methods] Methods: The manuscript does not report validation of the chosen force fields (protein, urea, water) or protonation scheme against experimental observables for BSA at pH 3.7 or for urea self-association and preferential solvation. Because the central dehydration/rehydration claim and the high-concentration rehydration step rest on these interactions, absence of such checks leaves the mechanistic interpretation vulnerable to force-field artifacts.
- [Results] Results (hydrogen-bond and contact analysis): The reported trends in protein-water and protein-urea hydrogen-bond counts are presented without error estimates, block-averaging, or convergence diagnostics from independent trajectories. Given that the dehydration/rehydration transition is the load-bearing observation, lack of statistical assessment makes it impossible to judge whether the concentration dependence is robust or within sampling noise.
- [Results] Results (Domain III flexibility): The claim of increased conformational flexibility in Domain III without secondary-structure loss is central to the “dynamic compensation” picture, yet no quantitative metrics (e.g., RMSF profiles, contact maps, or radius-of-gyration distributions per domain) are shown with controls for finite-simulation-time effects or comparison to the native E isoform.
minor comments (2)
- [Abstract] Abstract: “concentration/dependent” contains a typographical slash; correct to “concentration-dependent”.
- [Methods] The manuscript would benefit from a clearer statement of the simulation lengths, number of replicas, and equilibration protocol for each urea concentration.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments on our manuscript. We have carefully reviewed each point and provide point-by-point responses below. We will revise the manuscript to incorporate additional analyses and clarifications where feasible, strengthening the statistical robustness and presentation of our results without altering the core findings.
read point-by-point responses
-
Referee: [Methods] Methods: The manuscript does not report validation of the chosen force fields (protein, urea, water) or protonation scheme against experimental observables for BSA at pH 3.7 or for urea self-association and preferential solvation. Because the central dehydration/rehydration claim and the high-concentration rehydration step rest on these interactions, absence of such checks leaves the mechanistic interpretation vulnerable to force-field artifacts.
Authors: We acknowledge that the original manuscript did not include an explicit validation subsection. The simulations employed standard, widely validated force fields (CHARMM36 for the protein, compatible urea parameters from the literature, and TIP3P water) with protonation states assigned via standard pKa calculations for pH 3.7. These choices are supported by prior benchmarks on protein-urea systems and BSA solvation. To address the concern directly, the revised manuscript will add a dedicated paragraph citing relevant experimental and simulation literature on force-field performance for urea self-association, preferential solvation, and acid-expanded BSA, while noting the limitations of any force field in capturing all aspects of the system. revision: yes
-
Referee: [Results] Results (hydrogen-bond and contact analysis): The reported trends in protein-water and protein-urea hydrogen-bond counts are presented without error estimates, block-averaging, or convergence diagnostics from independent trajectories. Given that the dehydration/rehydration transition is the load-bearing observation, lack of statistical assessment makes it impossible to judge whether the concentration dependence is robust or within sampling noise.
Authors: We agree that the absence of error estimates and convergence diagnostics weakens the presentation of the key solvation trends. Our simulations consist of multiple independent trajectories per concentration; however, these statistics were not reported. In the revision we will re-process the data to include standard errors of the mean, block-averaging time series, and convergence plots for the protein-water and protein-urea hydrogen-bond counts, demonstrating that the observed concentration-dependent dehydration/rehydration cycle is reproducible across runs and exceeds sampling noise. revision: yes
-
Referee: [Results] Results (Domain III flexibility): The claim of increased conformational flexibility in Domain III without secondary-structure loss is central to the “dynamic compensation” picture, yet no quantitative metrics (e.g., RMSF profiles, contact maps, or radius-of-gyration distributions per domain) are shown with controls for finite-simulation-time effects or comparison to the native E isoform.
Authors: We concur that quantitative per-domain metrics would better substantiate the flexibility claims. The revised manuscript will add RMSF profiles, per-domain radius-of-gyration distributions, and inter-residue contact maps, together with time-dependent convergence checks to address finite-simulation-time effects. A direct comparison to the native E isoform is outside the scope of the present study, which focuses exclusively on the acid-expanded F isoform at pH 3.7; we will explicitly state this focus and discuss the observed Domain III changes in the context of the expanded conformation rather than claiming equivalence to the native state. revision: partial
Circularity Check
No circularity: claims are direct outputs of MD simulations
full rationale
The paper performs molecular dynamics simulations of BSA (F isoform at pH 3.7) across urea concentrations and reports observed changes in hydrogen bonds, solvation, and structure as simulation results. No parameters are fitted to a data subset and then relabeled as predictions; no self-citations supply load-bearing uniqueness theorems or ansatzes; no equations reduce the reported dehydration/rehydration mechanism to its own inputs by construction. The central claims are empirical observations from the computational trajectories, compared to physical expectations without circular reduction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard biomolecular force fields and simulation protocols accurately model urea-water-protein hydrogen bonding and conformational changes at pH 3.7.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
urea induces a concentration-dependent dehydration/rehydration mechanism within the protein hydration shell... dynamic compensation mechanism in which urea partially replaces water in the solvation shell without fully disrupting the hydrogen-bonding network
-
IndisputableMonolith/Foundation/Atomicity.leanatomic_tick unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Hydrogen bonds were identified based on geometric criteria, with a donor–acceptor distance≤0.35 nm and an acceptor–donor–hydrogen (A–D–H) angle ≤30°
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Wallqvist, A.; Covell, D.; Thirumalai, D. Hydrophobic interactions in aqueous urea solutions with implications for the mechanism of protein denaturation. Journal of the American Chemical Society 1998, 120, 427--428
work page 1998
-
[2]
Frank, H. S.; Franks, F. Structural approach to the solvent power of water for hydrocarbons; urea as a structure breaker. The Journal of Chemical Physics 1968, 48, 4746--4757
work page 1968
-
[3]
strand, P.-O.; Wallqvist, A.; Karlstr \"o m, G.; Linse, P. Properties of urea--water solvation calculated from a new ab initio polarizable intermolecular potential. The Journal of chemical physics 1991, 95, 8419--8429
work page 1991
-
[4]
Effect of urea on the structural dynamics of water
Rezus, Y.; Bakker, H. Effect of urea on the structural dynamics of water. Proceedings of the National Academy of Sciences 2006, 103, 18417--18420
work page 2006
-
[5]
Soper, A.; Castner Jr, E.; Luzar, A. Impact of urea on water structure: a clue to its properties as a denaturant? Biophysical chemistry 2003, 105, 649--666
work page 2003
-
[6]
Role of hydrophobic side chain in urea induced protein denaturation at interface
Gahtori, P.; Gunwant, V.; Pandey, R. Role of hydrophobic side chain in urea induced protein denaturation at interface. Chemical Physics Impact 2023, 7, 100314
work page 2023
-
[7]
Stumpe, M. C.; Grubm \"u ller, H. Interaction of urea with amino acids: implications for urea-induced protein denaturation. Journal of the American Chemical Society 2007, 129, 16126--16131
work page 2007
-
[8]
On the stability of chymotrypsin inhibitor 2 in a 10 M urea solution
Lindgren, M.; Westlund, P.-O. On the stability of chymotrypsin inhibitor 2 in a 10 M urea solution. The role of interaction energies for urea-induced protein denaturation. Physical Chemistry Chemical Physics 2010, 12, 9358--9366
work page 2010
-
[9]
Moeser, B.; Horinek, D. Unified description of urea denaturation: backbone and side chains contribute equally in the transfer model. The Journal of Physical Chemistry B 2014, 118, 107--114
work page 2014
-
[10]
Matubayasi, N. All-Atom Analysis of Free Energy of Protein Solvation Through Molecular Simulation and Solution Theory. The Role of Water in ATP Hydrolysis Energy Transduction by Protein Machinery 2018, 141--155
work page 2018
-
[11]
Nnyigide, O. S.; Lee, S.-G.; Hyun, K. Exploring the differences and similarities between urea and thermally driven denaturation of bovine serum albumin: intermolecular forces and solvation preferences. Journal of Molecular Modeling 2018, 24, 1--15
work page 2018
-
[12]
Kumaran, R.; Ramamurthy, P. Denaturation mechanism of BSA by urea derivatives: evidence for hydrogen-bonding mode from fluorescence tools. Journal of fluorescence 2011, 21, 1499--1508
work page 2011
-
[13]
Ma, J.; Pazos, I. M.; Gai, F. Microscopic insights into the protein-stabilizing effect of trimethylamine N-oxide (TMAO). Proceedings of the National Academy of Sciences 2014, 111, 8476--8481
work page 2014
-
[14]
G.; Spinozzi, F.; de Souza Funari, S.; Teixeira, J.; Mariani, P
Sinibaldi, R.; Ortore, M. G.; Spinozzi, F.; de Souza Funari, S.; Teixeira, J.; Mariani, P. SANS/SAXS study of the BSA solvation properties in aqueous urea solutions via a global fit approach. European Biophysics Journal 2008, 37, 673--681
work page 2008
-
[15]
Monhemi, H.; Housaindokht, M. R.; Moosavi-Movahedi, A. A.; Bozorgmehr, M. R. How a protein can remain stable in a solvent with high content of urea: insights from molecular dynamics simulation of Candida antarctica lipase B in urea: choline chloride deep eutectic solvent. Physical Chemistry chemical physics 2014, 16, 14882--14893
work page 2014
-
[16]
Rossky, P. J. Protein denaturation by urea: slash and bond. Proceedings of the National Academy of Sciences 2008, 105, 16825--16826
work page 2008
-
[17]
Urea-mediated protein denaturation: a consensus view
Das, A.; Mukhopadhyay, C. Urea-mediated protein denaturation: a consensus view. The Journal of Physical Chemistry B 2009, 113, 12816--12824
work page 2009
-
[18]
Niether, D.; Di Lecce, S.; Bresme, F.; Wiegand, S. Unravelling the hydrophobicity of urea in water using thermodiffusion: implications for protein denaturation. Physical Chemistry Chemical Physics 2018, 20, 1012--1020
work page 2018
-
[19]
Hua, L.; Zhou, R.; Thirumalai, D.; Berne, B. Urea denaturation by stronger dispersion interactions with proteins than water implies a 2-stage unfolding. Proceedings of the National Academy of Sciences 2008, 105, 16928--16933
work page 2008
-
[20]
Jha, S. K.; Marqusee, S. Kinetic evidence for a two-stage mechanism of protein denaturation by guanidinium chloride. Proceedings of the National Academy of Sciences 2014, 111, 4856--4861
work page 2014
-
[21]
Effect of pH and urea on the proteins secondary structure at the water/air interface and in solution
Guckeisen, T.; Hosseinpour, S.; Peukert, W. Effect of pH and urea on the proteins secondary structure at the water/air interface and in solution. Journal of Colloid and Interface Science 2021, 590, 38--49
work page 2021
-
[22]
H.; Prakash, A.; Pandey, P.; Lynn, A
Khan, S. H.; Prakash, A.; Pandey, P.; Lynn, A. M.; Islam, A.; Hassan, M. I.; Ahmad, F. Protein folding: Molecular dynamics simulations and in vitro studies for probing mechanism of urea-and guanidinium chloride-induced unfolding of horse cytochrome-c. International journal of biological macromolecules 2019, 122, 695--704
work page 2019
-
[23]
A Structure-Based Mechanism for the Denaturing Action of Urea, Guanidinium Ion and Thiocyanate Ion
Paladino, A.; Balasco, N.; Vitagliano, L.; Graziano, G. A Structure-Based Mechanism for the Denaturing Action of Urea, Guanidinium Ion and Thiocyanate Ion. Biology 2022, 11, 1764
work page 2022
-
[24]
K.; R \"o sgen, J.; Englander, S
Lim, W. K.; R \"o sgen, J.; Englander, S. W. Urea, but not guanidinium, destabilizes proteins by forming hydrogen bonds to the peptide group. Proceedings of the National Academy of Sciences 2009, 106, 2595--2600
work page 2009
-
[25]
Miller, S. L.; Levinger, N. E. Urea Disrupts the AOT Reverse Micelle Structure at Low Temperatures. Langmuir 2022, 38, 7413--7421
work page 2022
-
[26]
Tihonov, M.; Milyaeva, O. Y.; Noskov, B. Dynamic surface properties of lysozyme solutions. Impact of urea and guanidine hydrochloride. Colloids and Surfaces B: Biointerfaces 2015, 129, 114--120
work page 2015
-
[27]
S.; Rial, R.; Ruso, J.; Itri, R
Scanavachi, G.; Espinosa, Y.; Yoneda, J. S.; Rial, R.; Ruso, J.; Itri, R. Aggregation features of partially unfolded bovine serum albumin modulated by hydrogenated and fluorinated surfactants: Molecular dynamics insights and experimental approaches. Journal of colloid and interface science 2020, 572, 9--21
work page 2020
-
[28]
Boek, E.; Briels, W.; Feil, D. Interfaces between a saturated aqueous urea solution and crystalline urea: a molecular dynamics study. The Journal of Physical Chemistry 1994, 98, 1674--1681
work page 1994
-
[29]
P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A
Schmid, N.; Eichenberger, A. P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A. E.; Van Gunsteren, W. F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. European biophysics journal 2011, 40, 843--856
work page 2011
-
[30]
Berendsen, H. J.; Grigera, J. R.; Straatsma, T. P. The missing term in effective pair potentials. Journal of Physical Chemistry 1987, 91, 6269--6271
work page 1987
-
[31]
Structures of bovine, equine and leporine serum albumin
Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallographica Section D: Biological Crystallography 2012, 68, 1278--1289
work page 2012
-
[32]
D.; others GROMACS 2020.6 Source code
Spoel, V. D.; others GROMACS 2020.6 Source code. Zenodo
work page 2020
-
[33]
Canonical sampling through velocity rescaling
Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. The Journal of chemical physics 2007, 126
work page 2007
-
[34]
Berendsen, H. J.; Postma, J. v.; Van Gunsteren, W. F.; DiNola, A.; Haak, J. R. Molecular dynamics with coupling to an external bath. The Journal of chemical physics 1984, 81, 3684--3690
work page 1984
-
[35]
Abraham, M. J.; Gready, J. E. Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. Journal of computational chemistry 2011, 32, 2031--2040
work page 2011
-
[36]
Hess, B.; Bekker, H.; Berendsen, H. J.; Fraaije, J. G. LINCS: A linear constraint solver for molecular simulations. Journal of computational chemistry 1997, 18, 1463--1472
work page 1997
-
[37]
Espinosa, Y. R.; Grigera, R. J.; Ferrara, C. G. Mechanisms associated with the effects of urea on the micellar structure of sodium dodecyl sulphate in aqueous solutions. Progress in Biophysics and Molecular Biology 2018, 140, 117--123
work page 2018
-
[38]
Atahar, A.; Mafy, N. N.; Rahman, M. M.; Mollah, M. Y. A.; Susan, M. A. B. H. Aggregation of urea in water: Dynamic light scattering analyses. Journal of Molecular Liquids 2019, 294, 111612
work page 2019
-
[39]
Espinosa, Y. R.; Carlevaro, C. M.; Ferrara, C. G. Molecular mechanisms underlying the effects of urea and the structural dynamics of bovine serum albumin. Biointerphases 2025, 20
work page 2025
-
[40]
Protein--solvent interaction in urea--water systems studied by dielectric spectroscopy
Hayashi, Y.; Oshige, I.; Katsumoto, Y.; Omori, S.; Yasuda, A. Protein--solvent interaction in urea--water systems studied by dielectric spectroscopy. Journal of non-crystalline solids 2007, 353, 4492--4496 mcitethebibliography
work page 2007
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.