Recognition: 2 theorem links
· Lean TheoremVibeProteinBench: An Evaluation Benchmark for Language-interfaced Vibe Protein Design
Pith reviewed 2026-05-13 01:04 UTC · model grok-4.3
The pith
No large language model performs strongly across all stages of language-based protein design.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
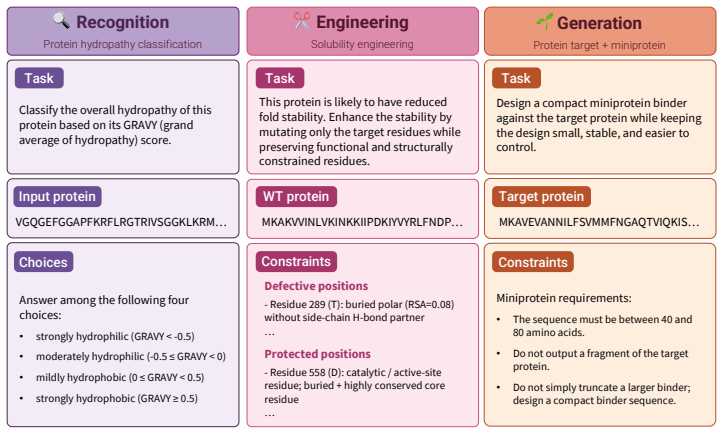
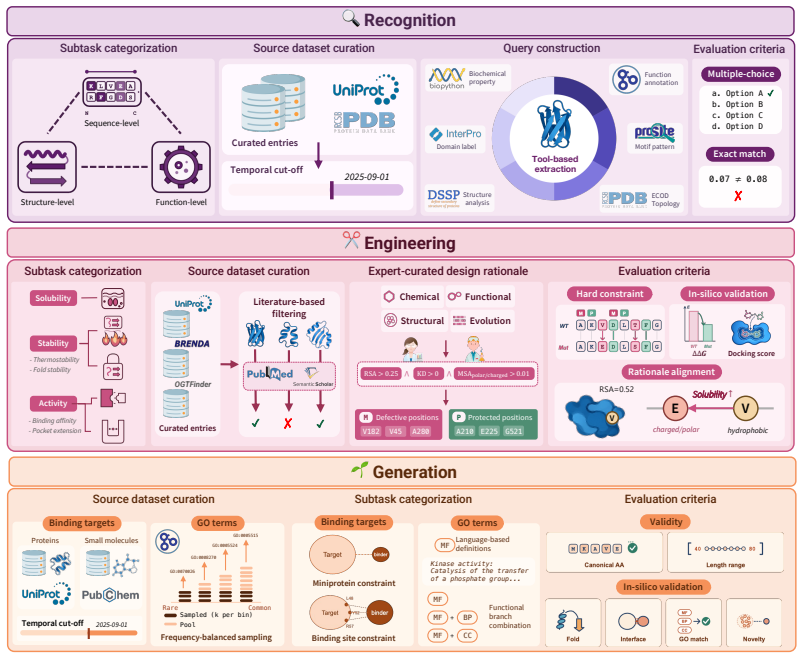
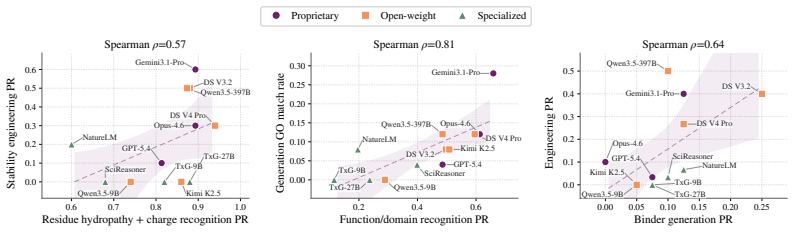
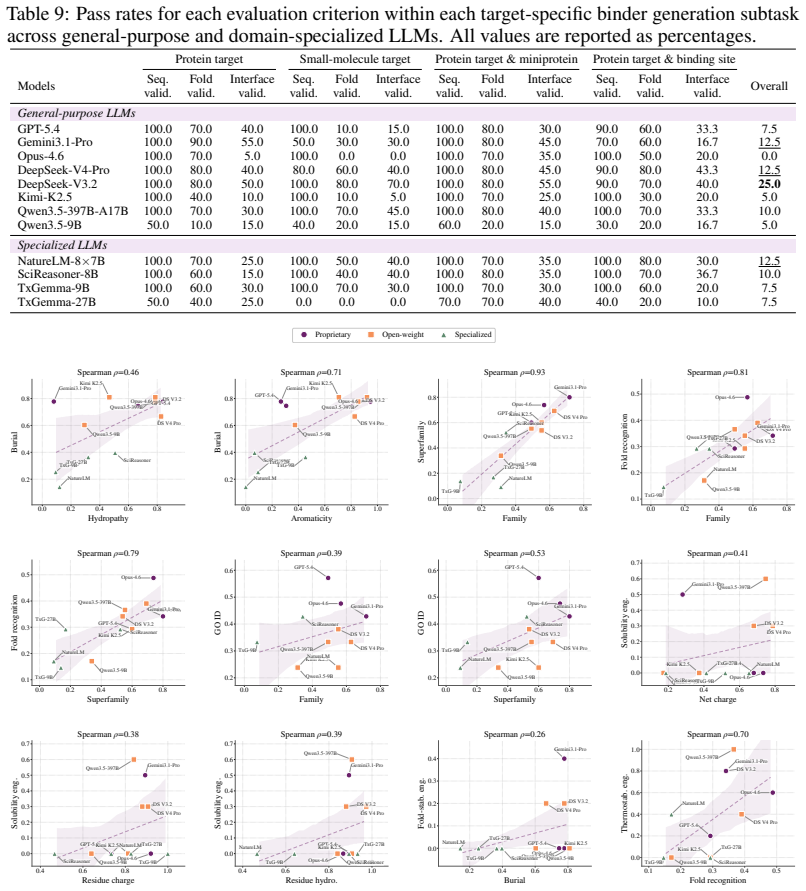
VibeProteinBench is a language-interfaced evaluation that covers the complete protein design workflow through three stages: recognition of properties from text, engineering of sequences under constraints, and generation of novel sequences. Each stage is supported by expert-curated mechanistic rationales and multi-faceted in silico validation to assess biological plausibility. Tests across general-purpose and domain-specialized LLMs show that no single model achieves strong results in all three stages at once, establishing that generalist protein design remains a substantial open challenge for current LLMs.
What carries the argument
VibeProteinBench, a three-stage benchmark (recognition, engineering, generation) that evaluates language-interfaced protein design using expert rationales and in silico checks to confirm biological plausibility of outputs.
If this is right
- Effective LLM-based protein design requires models that can handle recognition, engineering, and generation as an integrated process rather than isolated skills.
- Both general-purpose and specialized LLMs currently exhibit complementary weaknesses that prevent strong performance across the full workflow.
- The benchmark supplies a repeatable, language-based method for measuring future improvements in LLM capabilities for protein tasks.
- Progress toward generalist protein design will depend on closing gaps that appear when models must reason about and produce sequences under flexible natural-language goals.
Where Pith is reading between the lines
- Future work could extend the benchmark by adding real experimental feedback loops to test whether high in silico scores predict actual lab success.
- The results suggest that hybrid systems combining LLMs with physics-based modeling tools may be needed to overcome the current performance ceiling on integrated tasks.
- This evaluation framework could be adapted to other biological design domains that rely on natural language instructions, such as small-molecule or RNA design.
Load-bearing premise
Expert-curated mechanistic rationales together with computer simulations are sufficient to judge whether model-generated proteins are biologically plausible.
What would settle it
A controlled lab experiment that synthesizes and functionally tests proteins designed by a model that scored high on the benchmark, to see if the designs actually work as the in silico checks predicted.
Figures

read the original abstract
Protein design aims to compose amino-acid sequences that fold into stable three-dimensional structures while satisfying targeted functional properties. The field is increasingly shifting toward vibe protein design, where a single model is expected to generate novel sequences, engineer existing proteins, and reason about protein characteristics through flexible natural-language constraints. Large language models (LLMs) have emerged as a leading paradigm in this space. However, existing evaluation benchmarks often limit their scope to a partial aspect of protein design, while others restrict design objectives to structured input schemas, lacking an integrated framework that evaluates the broad spectrum of protein design competence under open-ended intents. To this end, we present Vibe Protein design Benchmark (VibeProteinBench), a language-interfaced benchmark that probes generalist capabilities through three complementary stages mirroring a computational protein design workflow: recognition, engineering, and generation. Each stage is grounded in expert-curated mechanistic rationales and multi-faceted in silico validation, to computationally verify whether model outputs are biologically plausible. Evaluations across diverse general-purpose and domain-specialized LLMs reveal that no model achieves strong performance across all three stages, suggesting that generalist protein design remains a substantial open challenge for current LLMs.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces VibeProteinBench, a language-interfaced benchmark for evaluating LLMs on protein design under open-ended natural language inputs. It comprises three stages—recognition, engineering, and generation—each grounded in expert-curated mechanistic rationales and multi-faceted in silico validation (structure prediction, stability, functional motif checks) to computationally assess biological plausibility of outputs. Evaluations across general-purpose and domain-specialized LLMs find that no model achieves strong performance across all stages, concluding that generalist protein design remains a substantial open challenge.
Significance. If the in silico validation protocols reliably indicate biological plausibility, the benchmark would be significant for providing an integrated, flexible framework that goes beyond partial or schema-restricted prior evaluations, directly testing LLM capabilities on realistic design workflows. The work is strengthened by testing external models with no reduction to self-fitted parameters or circular derivations from prior author results. However, the conditional nature of the significance (tied to unverified computational proxies) tempers its immediate impact on the field.
major comments (2)
- [Abstract] Abstract: The central claim that 'no model achieves strong performance across all three stages' and that 'generalist protein design remains a substantial open challenge' is load-bearing on the assumption that expert-curated mechanistic rationales plus multi-faceted in silico validation 'computationally verify whether model outputs are biologically plausible.' This mapping from computational filters to real-world plausibility is untested; common issues with de novo design (accepting non-functional sequences or rejecting viable ones) could systematically bias the 'no model succeeds' result without experimental calibration against known functional proteins.
- [Evaluation sections] Evaluation sections: The manuscript lacks specific reporting of metrics (e.g., exact scoring thresholds, R^{2} values, or success rates per stage), dataset sizes, curation details, and precise validation protocols (which structure predictors, stability metrics, motif databases, and decision rules are used). This absence makes it impossible to reproduce the benchmark or rule out curation bias, directly affecting confidence in the cross-stage performance comparisons.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript introducing VibeProteinBench. We address the major comments point-by-point below, providing clarifications and committing to revisions where appropriate to improve reproducibility and acknowledge limitations.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that 'no model achieves strong performance across all three stages' and that 'generalist protein design remains a substantial open challenge' is load-bearing on the assumption that expert-curated mechanistic rationales plus multi-faceted in silico validation 'computationally verify whether model outputs are biologically plausible.' This mapping from computational filters to real-world plausibility is untested; common issues with de novo design (accepting non-functional sequences or rejecting viable ones) could systematically bias the 'no model succeeds' result without experimental calibration against known functional proteins.
Authors: We acknowledge the referee's concern regarding the reliance on untested computational proxies for biological plausibility. While our benchmark uses established in silico methods grounded in expert-curated rationales, we agree that without experimental calibration, there is potential for bias in assessing model performance. In the revised manuscript, we will update the abstract to more precisely state that the validation 'computationally assesses' plausibility and add a dedicated limitations paragraph discussing the strengths and potential shortcomings of the in silico filters, including references to known issues in de novo design validation. We will also provide additional justification for the chosen validation approaches based on literature. Note that performing new experimental validations is outside the scope of this work. revision: partial
-
Referee: [Evaluation sections] Evaluation sections: The manuscript lacks specific reporting of metrics (e.g., exact scoring thresholds, R^{2} values, or success rates per stage), dataset sizes, curation details, and precise validation protocols (which structure predictors, stability metrics, motif databases, and decision rules are used). This absence makes it impossible to reproduce the benchmark or rule out curation bias, directly affecting confidence in the cross-stage performance comparisons.
Authors: We thank the referee for highlighting the need for greater specificity in reporting. This is a fair criticism, and we will revise the manuscript to include comprehensive details. Specifically, we will add tables and text describing dataset sizes for each stage, the curation process for mechanistic rationales, exact success metrics and thresholds (including any R^{2} values or success rates), and full validation protocols specifying the tools used (e.g., particular structure predictors, stability calculation methods, motif databases, and the logical decision rules for determining plausibility). These additions will be placed in the main text or a new supplementary section to ensure reproducibility. revision: yes
- Experimental calibration of the in silico validation protocols using known functional proteins, which would require wet-lab experiments not included in this computational benchmark study.
Circularity Check
No circularity: new benchmark evaluated empirically on external models
full rationale
The paper introduces VibeProteinBench as a novel three-stage evaluation framework (recognition, engineering, generation) for LLM-based protein design. Each stage is defined with expert-curated mechanistic rationales and multi-faceted in silico checks, then applied to a range of general-purpose and domain-specialized LLMs. The central claim—that no model achieves strong performance across all stages—follows directly from these independent empirical results rather than any self-referential fitting, parameter renaming, or self-citation chain. The benchmark construction and validation steps are self-contained against external models and do not reduce the reported outcomes to the inputs by definition.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Expert-curated mechanistic rationales accurately capture biological mechanisms for grounding benchmark tasks
- domain assumption Multi-faceted in silico validation tools can determine biological plausibility of generated or engineered sequences
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclearno model achieves strong performance across all three stages
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.