Recognition: no theorem link
Deep Learning for Protein Complex Prediction and Design
Pith reviewed 2026-05-13 03:48 UTC · model grok-4.3
The pith
Domain-specific deep learning architectures and sequence-space search algorithms improve protein complex structure prediction and enable protein sequence design.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Domain-specific deep learning architectures that capture the hierarchical nature of protein structures, together with search algorithms that navigate sequence spaces to identify interacting homologs, improve complex structure prediction and enable protein sequence design.
What carries the argument
Domain-specific deep learning architectures capturing the hierarchical nature of protein structures and search algorithms navigating sequence spaces to identify interacting homologs.
If this is right
- More accurate predictions of how multiple proteins assemble into complexes.
- New ability to design protein sequences that form desired complexes.
- Better computational support for studying cellular functions involving protein interactions.
- Practical routes toward designing proteins for therapeutic applications.
Where Pith is reading between the lines
- The approach could speed up identification of protein targets for drugs by simulating interactions more reliably.
- Combining these models with lab experiments might allow faster validation of designed sequences.
- Hierarchical modeling ideas could extend to other multi-molecule systems such as nucleic acid complexes.
Load-bearing premise
That domain-specific architectures can meaningfully capture protein hierarchy and that the search algorithms can efficiently locate interacting homologs in vast sequence spaces without prohibitive cost or false positives.
What would settle it
A benchmark test on known protein complexes where the new architectures and search methods fail to outperform standard deep learning predictors in accuracy or speed.
Figures







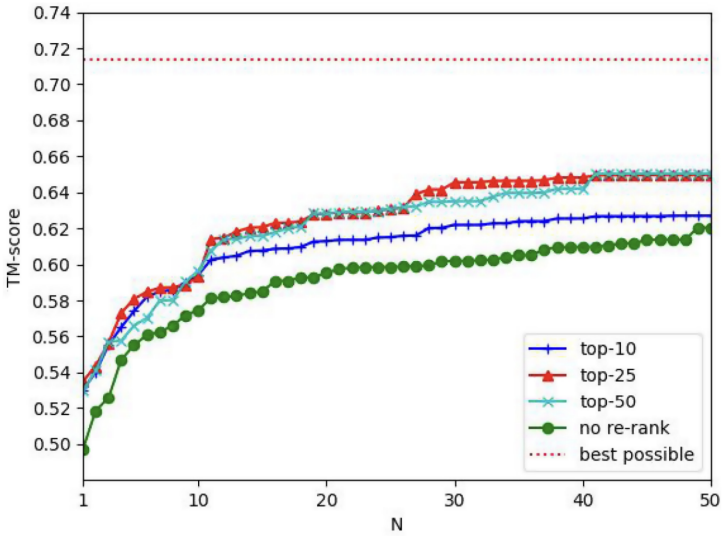

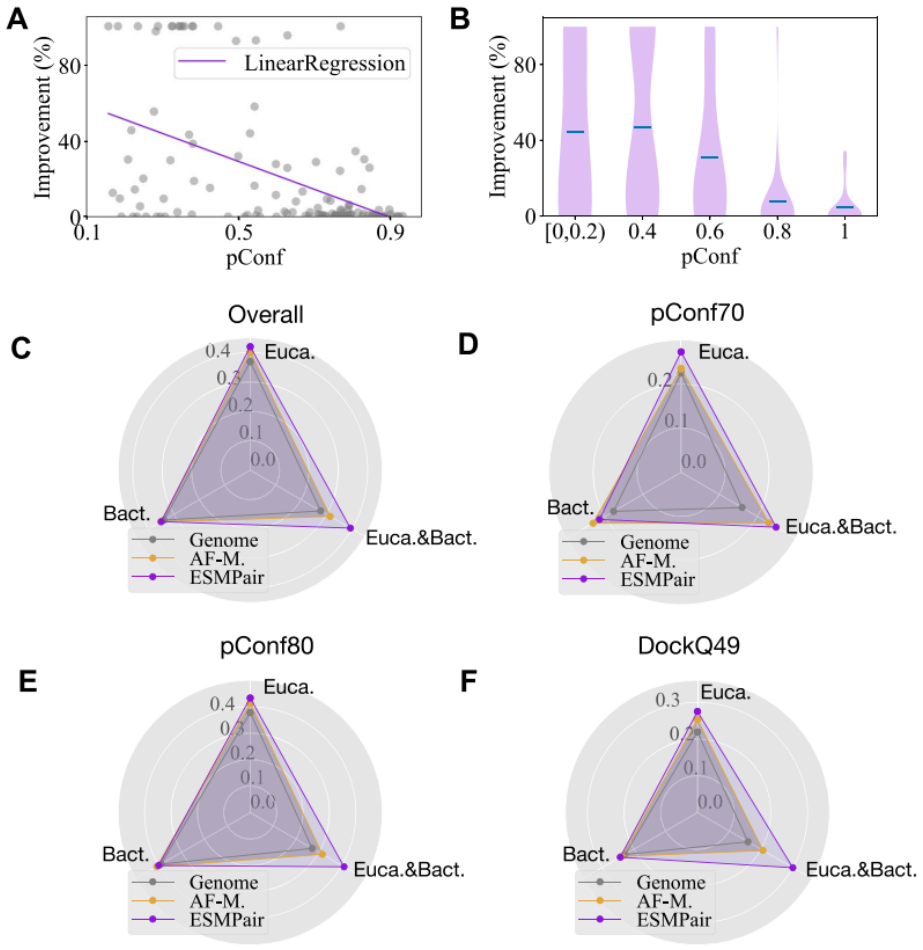






read the original abstract
Accurately modeling and designing protein complex structures is a central problem in computational structural biology, with broad implications for understanding cellular function and developing therapeutics. This thesis investigates two fundamental aspects of this problem using deep learning: domain-specific architectures that capture the hierarchical nature of protein structures, and search algorithms that efficiently navigate the vast sequence spaces of protein complexes to identify interacting homologs for improving complex structure prediction and to design protein sequences.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript is a thesis abstract claiming that domain-specific deep learning architectures capturing the hierarchical nature of protein structures, combined with search algorithms for navigating sequence spaces to identify interacting homologs, improve protein complex structure prediction and enable protein sequence design. No specific architectures, algorithms, datasets, loss functions, benchmarks, or results are described.
Significance. The topic addresses a central problem in computational structural biology with potential implications for cellular function understanding and therapeutics development. However, the absence of any methods, experimental validation, baselines, or quantitative results means the significance of the claimed improvements cannot be assessed; the contribution remains at the level of a high-level research plan rather than a demonstrated advance.
major comments (2)
- [Abstract] Abstract: The central claim that the proposed architectures and search algorithms 'improve complex structure prediction' and 'enable protein sequence design' is stated without any supporting methods, data, models, or results. This is load-bearing because the thesis's value rests entirely on demonstrating these improvements, yet no equations, architectures (e.g., no mention of specific layers or hierarchies), search procedures, or evaluation metrics are provided.
- No methods or results sections: The manuscript provides no description of training data, baselines (e.g., AlphaFold-Multimer or other complex predictors), performance metrics, or ablation studies. Without these, it is impossible to evaluate whether the domain-specific designs capture hierarchy better than existing approaches or whether the search algorithms scale without prohibitive cost or false positives.
Simulated Author's Rebuttal
We thank the referee for their review of our thesis abstract. We appreciate the feedback on the level of detail provided and address the major comments point by point below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that the proposed architectures and search algorithms 'improve complex structure prediction' and 'enable protein sequence design' is stated without any supporting methods, data, models, or results. This is load-bearing because the thesis's value rests entirely on demonstrating these improvements, yet no equations, architectures (e.g., no mention of specific layers or hierarchies), search procedures, or evaluation metrics are provided.
Authors: The manuscript is submitted as a thesis abstract, which by design offers a concise, high-level overview of the research program rather than a complete technical description. The domain-specific architectures for capturing protein structural hierarchy and the sequence-space search algorithms for identifying interacting homologs are developed and evaluated in the full thesis, including all equations, layer specifications, search procedures, datasets, and metrics. The abstract summarizes the motivation and claimed outcomes without repeating those details. We agree that a standalone research paper would require the supporting elements noted, but this submission follows the standard format for a thesis abstract. revision: no
-
Referee: [—] No methods or results sections: The manuscript provides no description of training data, baselines (e.g., AlphaFold-Multimer or other complex predictors), performance metrics, or ablation studies. Without these, it is impossible to evaluate whether the domain-specific designs capture hierarchy better than existing approaches or whether the search algorithms scale without prohibitive cost or false positives.
Authors: We acknowledge that the submitted text contains no methods or results sections. This is because the document is the abstract of the thesis; the full methods (including training data, baselines such as AlphaFold-Multimer, metrics, ablation studies, and scaling analyses) appear in the dedicated chapters of the thesis itself. The abstract is not intended to stand alone as a methods/results paper. If the referee's expectation is for a complete research article, we note that the current submission is a thesis summary and therefore does not include those sections. revision: no
- Providing the specific architectures, algorithms, datasets, loss functions, benchmarks, or quantitative results, as none of these details are present in the submitted abstract manuscript.
Circularity Check
No significant circularity in derivation chain
full rationale
The abstract and context describe a high-level investigation into domain-specific deep learning for protein complex prediction and design, with no equations, derivations, fitted parameters presented as predictions, or self-citations that could reduce claims to inputs by construction. No load-bearing steps matching the enumerated circularity patterns are present or identifiable from the provided text. The central claims remain descriptive statements of research focus rather than self-referential results.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Y. Akiyama, Z. Zhang, M. Mirdita, M. Steinegger, and S. Ovchinnikov, Scaling down protein language modeling with msa pairformer, bioRxiv, (2025), pp. 2025– 08
work page 2025
-
[2]
N. R. Bennett, B. Coventry, I. Goreshnik, B. Huang, A. Allen, D. Vafea- dos, Y. P. Peng, J. Dauparas, M. Baek, L. Stewart, et al.,Improving de novo protein binder design with deep learning, Nature Communications, 14 (2023), p. 2625
work page 2023
-
[3]
N. R. Bennett, J. L. Watson, R. J. Ragotte, A. J. Borst, D. L. See, C. Weidle, R. Biswas, Y. Yu, E. L. Shrock, R. Ault, et al.,Atomically accurate de novo design of antibodies with rfdiffusion, Nature, 649 (2026), pp. 183–193. 114
work page 2026
-
[4]
A. Bhatnagar, S. Jain, J. Beazer, S. C. Curran, A. M. Hoffnagle, K. S. Ching, M. Martyn, S. Nayfach, J. A. Ruffolo, and A. Madani,Scaling unlocks broader generation and deeper functional understanding of proteins, bioRxiv, (2025), pp. 2025–04
work page 2025
-
[5]
S. E. Boyken, M. A. Benhaim, F. Busch, M. Jia, M. J. Bick, H. Choi, J. C. Klima, Z. Chen, C. Walkey, A. Mileant, et al.,De novo design of tunable, ph-driven conformational changes, Science, 364 (2019), pp. 658–664
work page 2019
-
[6]
B. Chen, X. Cheng, P. Li, Y.-a. Geng, J. Gong, S. Li, Z. Bei, X. Tan, B. Wang, X. Zeng, et al.,xtrimopglm: unified 100-billion-parameter pretrained transformer for deciphering the language of proteins, Nature Methods, 22 (2025), pp. 1028–1039
work page 2025
-
[7]
A. E. Chu, J. Kim, L. Cheng, et al.,An all-atom protein generative model, Proceedings of the National Academy of Sciences, 121 (2024), p. e2311500121
work page 2024
-
[8]
A. E. Chu, T. Lu, and P.-S. Huang,Sparks of function by de novo protein design, Nature biotechnology, 42 (2024), pp. 203–215
work page 2024
-
[9]
M. Chungyoun, J. Ruffolo, and J. Gray,Flab: Benchmarking deep learning methods for antibody fitness prediction, BioRxiv, (2024), pp. 2024–01
work page 2024
-
[10]
T. Dao, D. Fu, S. Ermon, A. Rudra, and C. Ré,Flashattention: Fast and memory-efficient exact attention with io-awareness, Advances in neural information processing systems, 35 (2022), pp. 16344–16359
work page 2022
-
[11]
J. Dauparas, I. Anishchenko, N. Bennett, et al.,Robust deep learning-based protein sequence design using proteinmpnn, Science, 378 (2022), pp. 49–56
work page 2022
-
[12]
H. Dieckhaus, M. Brocidiacono, N. Z. Randolph, and B. Kuhlman,Transfer learning to leverage larger datasets for improved prediction of protein stability changes, Proceedings of the national academy of sciences, 121 (2024), p. e2314853121. 115
work page 2024
- [13]
-
[14]
R. R. Eguchi, C. A. Choe, and P.-S. Huang,Ig-vae: Generative modeling of protein structure by direct 3d coordinate generation, PLoS computational biology, 18 (2022), p. e1010271
work page 2022
- [15]
- [16]
-
[17]
M. Gao, D. Nakajima An, J. M. Parks, and J. Skolnick,Af2complex predicts direct physical interactions in multimeric proteins with deep learning, Nature communi- cations, 13 (2022), p. 1744
work page 2022
- [18]
-
[19]
M. H. Høie, A. M. Hummer, T. H. Olsen, B. Aguilar-Sanjuan, M. Nielsen, and C. M. Deane,Antifold: Improved structure-based antibody design using inverse folding, Bioinformatics Advances, 5 (2025), p. vbae202
work page 2025
-
[20]
J. B. Ingraham, M. Baranov, Z. Costello, K. W. Barber, W. Wang, A. Ismail, V. Frappier, D. M. Lord, C. Ng-Thow-Hing, E. R. Van Vlack, et al.,Illuminating protein space with a programmable generative model, Nature, 623 (2023), pp. 1070–1078. 116
work page 2023
-
[21]
I. Johansson-Åkhe and B. Wallner,Improving peptide-protein docking with alphafold-multimer using forced sampling, Frontiers in bioinformatics, 2 (2022), p. 959160
work page 2022
- [22]
-
[23]
A. Mirarchi, T. Giorgino, and G. De Fabritiis,mdcath: A large-scale md dataset for data-driven computational biophysics, Scientific Data, 11 (2024), p. 1299
work page 2024
- [24]
-
[25]
E. Nijkamp, J. A. Ruffolo, E. N. Weinstein, N. Naik, and A. Madani, Progen2: exploring the boundaries of protein language models, Cell systems, 14 (2023), pp. 968–978
work page 2023
-
[26]
P. Notin, A. Kollasch, D. Ritter, L. Van Niekerk, S. Paul, H. Spinner, N. Rollins, A. Shaw, R. Orenbuch, R. Weitzman, et al.,Proteingym: Large- scale benchmarks for protein fitness prediction and design, Advances in neural information processing systems, 36 (2023), pp. 64331–64379
work page 2023
- [27]
- [28]
-
[29]
W. Qu, J. Guan, R. Ma, and K. Zhai,P(all-atom) is unlocking new path for protein design, bioRxiv, (2024)
work page 2024
-
[30]
J. P. Roney, C. Ou, and S. Ovchinnikov,Protein diffusion models as statistical potentials, bioRxiv, (2025), pp. 2025–12
work page 2025
-
[31]
J. P. Roney and S. Ovchinnikov,State-of-the-art estimation of protein model accuracy using alphafold, Physical review letters, 129 (2022), p. 238101
work page 2022
-
[32]
D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, et al.,Kemp elimination catalysts by computational enzyme design, Nature, 453 (2008), pp. 190–195
work page 2008
-
[33]
E.-M. Strauch, S. J. Fleishman, and D. Baker,Computational design of a ph-sensitive igg binding protein, Proceedings of the National Academy of Sciences, 111 (2014), pp. 675–680
work page 2014
-
[34]
J. Su, C. Han, Y. Zhou, J. Shan, X. Zhou, and F. Yuan,Saprot: Protein language modeling with structure-aware vocabulary, bioRxiv, (2023)
work page 2023
-
[35]
K. Tsuboyama, J. Dauparas, J. Chen, E. Laine, Y. Mohseni Behbahani, J. J. Weinstein, N. M. Mangan, S. Ovchinnikov, and G. J. Rocklin,Mega-scale experimental analysis of protein folding stability in biology and design, Nature, 620 (2023), pp. 434–444
work page 2023
-
[36]
J. L. Watson, D. Juergens, N. R. Bennett, B. L. Trippe, J. Yim, H. E. Eisenach, W. Ahern, A. J. Borst, R. J. Ragotte, L. F. Milles, et al., De novo design of protein structure and function with rfdiffusion, Nature, 620 (2023), pp. 1089–1100. 118
work page 2023
-
[37]
H. K. Wayment-Steele, A. Ojoawo, R. Otten, J. M. Apitz, W. Pitsawong, M. Hömberger, S. Ovchinnikov, L. Colwell, and D. Kern,Predicting multiple conformations via sequence clustering and AlphaFold2, Nature, 625 (2024), pp. 832–839
work page 2024
-
[38]
T. Widatalla, R. Rafailov, and B. Hie,Aligning protein generative models with experimental fitness via direct preference optimization, bioRxiv, (2024), pp. 2024–05
work page 2024
-
[39]
J. Zhou, C. Q. Le, Y. Zhang, and J. A. Wells,A general approach for selection of epitope-directed binders to proteins, Proceedings of the National Academy of Sciences, 121 (2024), p. e2317307121. 119
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.