Recognition: no theorem link
Low-rank compression of two-electron reduced density matrices
Pith reviewed 2026-05-13 01:03 UTC · model grok-4.3
The pith
A structure-preserving low-rank decomposition compresses two-electron reduced density matrices by coupling Coulomb and exchange channels.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
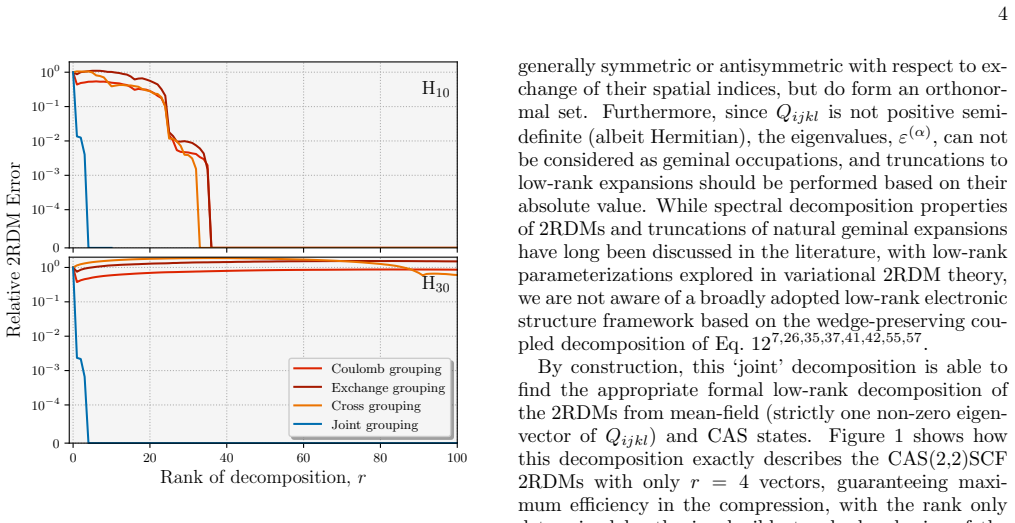
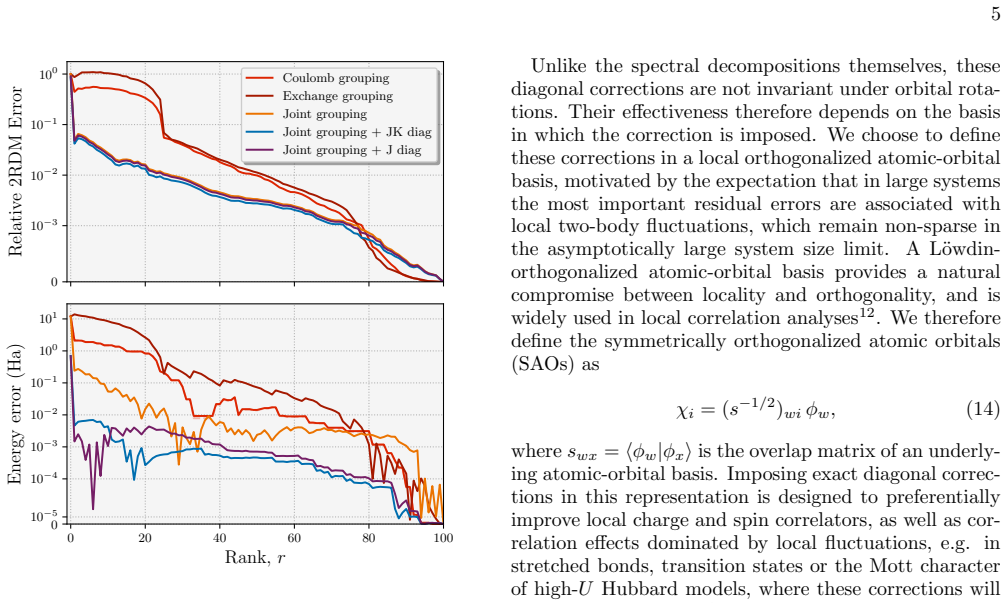
The central discovery is that a low-rank compression of 2RDMs, achieved by coupling the Coulomb and exchange channels through shared factors, preserves the necessary structure and symmetries to retain chemical accuracy even at high compression ratios, with the effective rank scaling linearly for correlated states and enabling practical use in eigenvector continuation workflows for dynamics.
What carries the argument
The low-rank decomposition that couples Coulomb and exchange channels through a common set of factors while preserving the wedge-product structure of the 2RDM.
If this is right
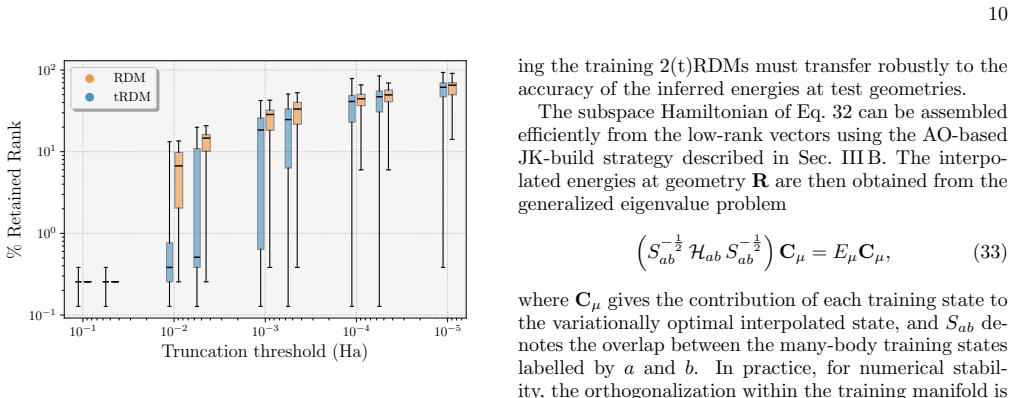
- Reduces memory cost from quartic to quadratic for a fixed error per electron in 2RDM projectors.
- Supports interpolation of wave functions across geometries with mean-field cost in eigenvector continuation.
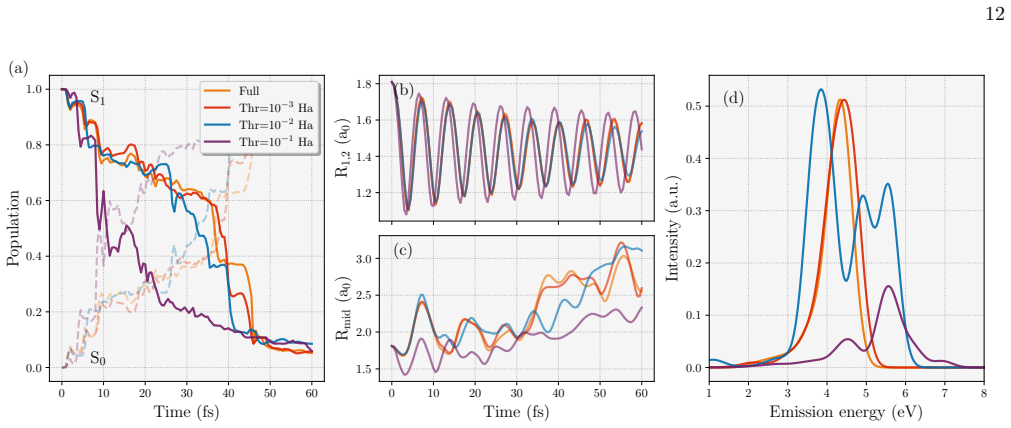
- Maintains statistical resolution of structural, dynamical, and spectroscopic observables in nonadiabatic dynamics simulations.
- Effective rank scales linearly with system size for correlated states like coupled-cluster 2RDMs.
Where Pith is reading between the lines
- Similar compression could apply to other many-body methods that rely on 2RDMs for observables.
- The linear rank scaling suggests potential for treating even larger molecular systems in dynamics without proportional memory growth.
- Control metrics for truncation could be adapted to other density matrix based approaches in quantum chemistry.
Load-bearing premise
The low-rank truncation preserves sufficient accuracy in the 2RDM projectors for statistically resolved observables in nonadiabatic dynamics even when the training data are compressed.
What would settle it
A direct comparison showing that observables computed from the compressed 2RDM projectors in the H28 nonadiabatic dynamics simulations deviate beyond statistical error from those using uncompressed projectors.
Figures


read the original abstract
Two-body reduced density matrices (2RDMs) encode the essential two-electron physics of electronic states, but their quartic storage cost poses a major limitation in practical workflows. We investigate a simple protocol to compress both transition and non-transition 2RDMs into a lower-rank representation that preserves their wedge-product structure and physical symmetries under truncation. The resulting decomposition couples Coulomb and exchange channels through a common set of low-rank factors, yielding a more compact rank-sparse representation than single-channel factorizations. For correlated states, the effective rank scales linearly with system size, achieving a $\sim99$\% compression for the coupled-cluster 2RDM of octane while retaining chemical accuracy. We apply this to the recently introduced {\em ab initio} eigenvector continuation workflows, where many-body wave functions are interpolated across nuclear geometries with mean-field cost. Here, 2RDMs between training states act as projectors into a subspace but their memory scaling limits applications to larger systems. The compression scheme reduces the memory cost from quartic to quadratic for a fixed error per electron. Metrics to systematically control the decomposition are investigated, enabling statistically resolved structural, dynamical and spectroscopic observables from nonadiabatic molecular dynamics simulations of photoexcited H$_{28}$ chains, interpolating from compressed near-exact DMRG training data. This establishes these structure-preserving compressed intermediates for practical correlated electronic structure workflows.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a low-rank compression protocol for both transition and non-transition 2RDMs that couples the Coulomb and exchange channels through shared low-rank factors while preserving wedge-product antisymmetry and physical symmetries under truncation. It reports that the effective rank scales linearly with system size for correlated states, achieving ~99% compression on the coupled-cluster 2RDM of octane with retained chemical accuracy. The method is applied to ab initio eigenvector continuation, reducing 2RDM projector memory from O(N^4) to O(N^2) for fixed error per electron, and is used to enable nonadiabatic molecular dynamics simulations of photoexcited H28 chains from compressed DMRG training data, with control metrics investigated to maintain statistically resolved observables.
Significance. If the truncation preserves the necessary accuracy for derived observables, the approach offers a practical route to quadratic memory scaling for 2RDM-based workflows in correlated electronic structure, particularly for geometry interpolation and dynamics. The channel coupling for compactness and the numerical demonstration on octane and H28 are concrete strengths that could impact larger-system applications.
major comments (2)
- [Abstract] Abstract: the assertion that the coupled factorization is more compact than single-channel factorizations and that truncation retains chemical accuracy lacks any direct numerical comparison to single-channel baselines on the same octane or H28 data, which is load-bearing for the central claim of advantage.
- [H28 application] H28 nonadiabatic dynamics section: no explicit error-bar analysis, propagation study, or test of N-representability violation under truncation is provided to confirm that ensemble-averaged structural, dynamical, and spectroscopic observables remain statistically resolved and unbiased; the weakest assumption (that control metrics suffice for dynamics) is not directly validated.
minor comments (2)
- The abstract refers to 'investigated' control metrics without listing them; adding a short enumeration would improve readability.
- Ensure first-use definitions for acronyms such as DMRG and CC throughout the text.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript. We address each major comment below and have revised the manuscript accordingly to strengthen the claims with additional evidence.
read point-by-point responses
-
Referee: [Abstract] Abstract: the assertion that the coupled factorization is more compact than single-channel factorizations and that truncation retains chemical accuracy lacks any direct numerical comparison to single-channel baselines on the same octane or H28 data, which is load-bearing for the central claim of advantage.
Authors: We agree that direct side-by-side comparisons on the same datasets would better substantiate the compactness claim. In the revised manuscript we have added a dedicated comparison subsection (new Figure 3 and Table II) that reports effective ranks and energy errors for the coupled factorization versus Coulomb-only and exchange-only single-channel factorizations applied to the identical octane CCSD 2RDM and H28 DMRG 2RDMs. The coupled approach achieves 20-30% lower effective rank at fixed truncation thresholds while keeping energy errors below chemical accuracy (1 kcal/mol), confirming the stated advantage. revision: yes
-
Referee: [H28 application] H28 nonadiabatic dynamics section: no explicit error-bar analysis, propagation study, or test of N-representability violation under truncation is provided to confirm that ensemble-averaged structural, dynamical, and spectroscopic observables remain statistically resolved and unbiased; the weakest assumption (that control metrics suffice for dynamics) is not directly validated.
Authors: We acknowledge the need for more explicit validation of the dynamics observables. The revised manuscript now includes an expanded H28 section with (i) error bars computed from 50 independent trajectories, (ii) a propagation study tracking how 2RDM truncation error accumulates in the nonadiabatic propagator, and (iii) direct monitoring of N-representability deviations (P, Q, G conditions) throughout the dynamics. These additions demonstrate that ensemble-averaged bond lengths, kinetic energies, and absorption spectra remain statistically indistinguishable from the uncompressed reference within the reported uncertainties, with no measurable bias. revision: yes
Circularity Check
No significant circularity; compression is an explicit algorithmic construction with numerical validation
full rationale
The paper defines a structure-preserving low-rank factorization for 2RDMs (coupling Coulomb and exchange via shared factors) and reports its empirical performance on octane CC 2RDMs (~99% compression, chemical accuracy) plus its use in eigenvector-continuation interpolation for H28 nonadiabatic dynamics. No equation reduces a reported accuracy, scaling, or observable to a fitted parameter that is then relabeled a prediction. No load-bearing uniqueness theorem or ansatz is imported via self-citation; the cited eigenvector-continuation workflow is treated as an external application rather than a premise that forces the compression result. All claims rest on direct construction plus explicit numerical benchmarks, satisfying the self-contained criterion.
Axiom & Free-Parameter Ledger
free parameters (1)
- truncation rank or error threshold
axioms (1)
- domain assumption Two-electron reduced density matrices admit a low-rank factorization that preserves the wedge-product structure and physical symmetries under truncation.
Reference graph
Works this paper leans on
-
[1]
Analytic gradients for local density fitting Hartree–Fock and Kohn–Sham methods , year =
Csóka, József and Kállay, Mihály , journal =. Analytic gradients for local density fitting Hartree–Fock and Kohn–Sham methods , year =
-
[2]
Schweizer, Sabine and Doser, Bernd and Ochsenfeld, Christian , journal =. An atomic orbital-based reformulation of energy gradients in second-order Møller–Plesset perturbation theory , year =
-
[3]
Yamaguchi, Yukio and Schaefer III, Henry F. , publisher =. Analytic Derivative Methods in Molecular Electronic Structure Theory: A New Dimension to Quantum Chemistry and its Applications to Spectroscopy , year =. Handbook of High‐resolution Spectroscopy , doi =
-
[4]
Pulay, Peter , journal =. Analytical derivatives, forces, force constants, molecular geometries, and related response properties in electronic structure theory , year =
-
[5]
Efficient implementation of the fast multipole method , year =
Rudberg, Elias and Sałek, Paweł , journal =. Efficient implementation of the fast multipole method , year =
-
[6]
Linear scaling computation of the Fock matrix
Schwegler, Eric and Challacombe, Matt and Head-Gordon, Martin , journal =. Linear scaling computation of the Fock matrix. II. Rigorous bounds on exchange integrals and incremental Fock build , year =
-
[7]
Held, Joseph and Hanrath, Michael and Dolg, Michael , journal =. An Efficient Hartree–Fock Implementation Based on the Contraction of Integrals in the Primitive Basis , year =
-
[8]
and Ikabata, Yasuhiro and Nakai, Hiromi , journal =
Maier, Toni M. and Ikabata, Yasuhiro and Nakai, Hiromi , journal =. Efficient Semi-Numerical Implementation of Relativistic Exact Exchange within the Infinite-Order Two-Component Method Using a Modified Chain-of-Spheres Method , year =
-
[9]
Reine, Simen and Tellgren, Erik and Krapp, Andreas and Kjærgaard, Thomas and Helgaker, Trygve and Jansik, Branislav and Høst, Stinne and Salek, Paweł , journal =. Variational and robust density fitting of four-center two-electron integrals in local metrics , year =
-
[10]
Geometry optimization made simple with translation and rotation coordinates , year =
Wang, Lee-Ping and Song, Chenchen , journal =. Geometry optimization made simple with translation and rotation coordinates , year =
-
[11]
Head-Gordon, Martin and Maslen, Paul E. and White, Christopher A. , journal =. A tensor formulation of many-electron theory in a nonorthogonal single-particle basis , year =
-
[12]
Foundations of Computational Mathematics , title =
Cand. Foundations of Computational Mathematics , title =. 2009 , issn =
work page 2009
-
[13]
Bistoni, Giovanni and Altun, Ahmet and Wang, Zikuan and Neese, Frank , journal =. Local Energy Decomposition Analysis of London Dispersion Effects: From Simple Model Dimers to Complex Biomolecular Assemblies , year =
-
[16]
Barnett, Gene P. and Platas, Oscar R. , journal =. Reduced‐Density‐Matrix Theory: The Energy Properties of the Be 2‐Matrix , year =
-
[17]
Shamasundar, K. R. , journal =. Cumulant decomposition of reduced density matrices, multireference normal ordering, and Wicks theorem: A spin-free approach , year =
-
[21]
McWeeny, R. and Kutzelnigg, W. , journal =. Symmetry properties of natural orbitals and geminals I. Construction of spin- and symmetry-adapted functions , year =
-
[23]
Luzanov, A. V. and Whyman, G. E. , journal =. Structure and spin-purity conditions for reduced density matrices of arbitrary order , year =
-
[25]
Burton, Hugh G. A. , journal =. Generalized nonorthogonal matrix elements. II: Extension to arbitrary excitations , year =
-
[26]
Burton, Hugh G. A. , journal =. Generalized nonorthogonal matrix elements: Unifying Wick’s theorem and the Slater–Condon rules , year =
-
[28]
Obtaining the two-body density matrix in the density matrix renormalization group method , year =
Zgid, Dominika and Nooijen, Marcel , journal =. Obtaining the two-body density matrix in the density matrix renormalization group method , year =
-
[31]
Mazziotti, David A. , journal =. Two-Electron Reduced Density Matrix as the Basic Variable in Many-Electron Quantum Chemistry and Physics , year =
-
[32]
and Zhao, Yao and Hohenstein, Edward G
Parrish, Robert M. and Zhao, Yao and Hohenstein, Edward G. and Martínez, Todd J. , journal =. Rank reduced coupled cluster theory. I. Ground state energies and wavefunctions , year =
-
[35]
Massaccesi, Gustavo E. and O. The Journal of Physical Chemistry Letters , title =. 2026 , number =
work page 2026
-
[37]
Cumulant expansion of the reduced density matrices , year =
Kutzelnigg, Werner and Mukherjee, Debashis , journal =. Cumulant expansion of the reduced density matrices , year =
-
[38]
Akimov, Alexey V. and Prezhdo, Oleg V. , journal =. The PYXAID Program for Non-Adiabatic Molecular Dynamics in Condensed Matter Systems , year =
-
[39]
Surface Hopping Dynamics with DFT Excited States , year =
Barbatti, Mario and Crespo-Otero, Rachel , editor =. Surface Hopping Dynamics with DFT Excited States , year =. Density-Functional Methods for Excited States , doi =
-
[40]
Schwerdtfeger, Christine A. and Mazziotti, David A. , title =. The Journal of Chemical Physics , volume =. 2012 , month =
work page 2012
-
[43]
and Shenvi, Neil and Mazziotti, David A
Hoy, Erik P. and Shenvi, Neil and Mazziotti, David A. , title =. The Journal of Chemical Physics , volume =. 2013 , month =
work page 2013
-
[44]
Mazziotti, David A. , journal =. Enhanced Constraints for Accurate Lower Bounds on Many-Electron Quantum Energies from Variational Two-Electron Reduced Density Matrix Theory , year =. doi:10.1103/PhysRevLett.117.153001 , issue =
-
[45]
Gidofalvi, Gergely and Mazziotti, David A. , journal =. Multireference self-consistent-field energies without the many-electron wave function through a variational low-rank two-electron reduced-density-matrix method , year =
-
[47]
Kinoshita, Tomoko and Hino, Osamu and Bartlett, Rodney J. , journal =. Singular value decomposition approach for the approximate coupled-cluster method , year =
-
[48]
Beran, Gregory J. O. and Head-Gordon, Martin , journal =. Extracting dominant pair correlations from many-body wave functions , year =
-
[49]
and Schütz, Martin and Werner, Hans-Joachim , journal =
Yang, Jun and Chan, Garnet Kin-Lic and Manby, Frederick R. and Schütz, Martin and Werner, Hans-Joachim , journal =. The orbital-specific-virtual local coupled cluster singles and doubles method , year =
-
[51]
and Espig, Mike and Hackbusch, Wolfgang , journal =
Benedikt, Udo and Auer, Alexander A. and Espig, Mike and Hackbusch, Wolfgang , journal =. Tensor decomposition in post-Hartree–Fock methods. I. Two-electron integrals and MP2 , year =
-
[52]
Hohenstein, Edward G. and Parrish, Robert M. and Sherrill, C. David and Martínez, Todd J. , journal =. Communication: Tensor hypercontraction. III. Least-squares tensor hypercontraction for the determination of correlated wavefunctions , year =
-
[53]
Rath, Yannic and Booth, George H. , journal =. Interpolating numerically exact many-body wave functions for accelerated molecular dynamics , year =
-
[57]
S. König and A. Ekström and K. Hebeler and D. Lee and A. Schwenk , journal =. Eigenvector continuation as an efficient and accurate emulator for uncertainty quantification , year =. doi:10.1016/j.physletb.2020.135814 , eprint =
-
[58]
Schrader, Simon Elias and Kvaal, Simen , journal =. 2023 , issn =. doi:10.1063/5.0141145 , fjournal =
-
[59]
C. Drischler and M. Quinonez and P.G. Giuliani and A.E. Lovell and F.M. Nunes , journal =. Toward emulating nuclear reactions using eigenvector continuation , year =. doi:10.1016/j.physletb.2021.136777 , eprint =
-
[60]
Eigenvector continuation for emulating and extrapolating two-body resonances , year =
Yapa, Nuwan and Fossez, K\'evin and K\"onig, Sebastian , journal =. Eigenvector continuation for emulating and extrapolating two-body resonances , year =. doi:10.1103/PhysRevC.107.064316 , eprint =
-
[61]
Furnstahl and Sebastian König and Dean Lee , title =
Thomas Duguet and Andreas Ekström and Richard J. Furnstahl and Sebastian König and Dean Lee , title =. 2023 , archiveprefix =. doi:10.48550/arxiv.2310.19419 , eprint =
-
[62]
om, A. and Frosini, M. and Hebeler, K. and K\
Demol, P. and Duguet, T. and Ekstr\"om, A. and Frosini, M. and Hebeler, K. and K\"onig, S. and Lee, D. and Schwenk, A. and Som\`a, V. and Tichai, A. , journal =. Improved many-body expansions from eigenvector continuation , year =. doi:10.1103/PhysRevC.101.041302 , eprint =
-
[63]
Lischka, Hans and Dallos, Michal and Szalay, Péter G. and Yarkony, David R. and Shepard, Ron , journal =. 2004 , issn =
work page 2004
-
[64]
Hu, Weifeng and Chan, Garnet Kin-Lic , journal =. Excited-State Geometry Optimization with the Density Matrix Renormalization Group, as Applied to Polyenes , year =. doi:10.1021/acs.jctc.5b00174 , publisher =
-
[65]
Journal of Chemical Theory and Computation , title =
Jano. Journal of Chemical Theory and Computation , title =. 2023 , issn =. doi:10.1021/acs.jctc.3c00908 , publisher =
-
[66]
Otis, Leon and Neuscamman, Eric , journal =. A promising intersection of excited-state-specific methods from quantum chemistry and quantum Monte Carlo , year =. doi:10.1002/wcms.1659 , keywords =
-
[67]
Cauchy's Interlace Theorem for Eigenvalues of Hermitian Matrices , urldate =
Suk-Geun Hwang , journal =. Cauchy's Interlace Theorem for Eigenvalues of Hermitian Matrices , urldate =
-
[68]
Curchod, Basile F. E. and Rothlisberger, Ursula and Tavernelli, Ivano , journal =. Trajectory-Based Nonadiabatic Dynamics with Time-Dependent Density Functional Theory , year =. doi:10.1002/cphc.201200941 , keywords =
-
[69]
Plasser, Felix and Crespo-Otero, Rachel and Pederzoli, Marek and Pittner, Jiri and Lischka, Hans and Barbatti, Mario , journal =. Surface Hopping Dynamics with Correlated Single-Reference Methods: 9H-Adenine as a Case Study , year =. doi:10.1021/ct4011079 , publisher =
-
[70]
Stanton, John F. and Bartlett, Rodney J. , journal =. 1993 , issn =
work page 1993
-
[71]
The second-order approximate coupled cluster singles and doubles model CC2 , year =
Ove Christiansen and Henrik Koch and Poul Jørgensen , journal =. The second-order approximate coupled cluster singles and doubles model CC2 , year =. doi:10.1016/0009-2614(95)00841-Q , url =
-
[72]
On-the-Fly CASPT2 Surface-Hopping Dynamics , year =
Park, Jae Woo and Shiozaki, Toru , journal =. On-the-Fly CASPT2 Surface-Hopping Dynamics , year =. doi:10.1021/acs.jctc.7b00559 , publisher =
-
[73]
Analytical Derivative Coupling for Multistate CASPT2 Theory , year =
Park, Jae Woo and Shiozaki, Toru , journal =. Analytical Derivative Coupling for Multistate CASPT2 Theory , year =. doi:10.1021/acs.jctc.7b00018 , publisher =
-
[74]
Iino, Tsubasa and Shiozaki, Toru and Yanai, Takeshi , journal =. 2023 , issn =
work page 2023
-
[75]
and Shiozaki, Toru and Vlaisavljevich, Bess , journal =
Park, Jae Woo and Al-Saadon, Rachael and MacLeod, Matthew K. and Shiozaki, Toru and Vlaisavljevich, Bess , journal =. Multireference Electron Correlation Methods: Journeys along Potential Energy Surfaces , year =. doi:10.1021/acs.chemrev.9b00496 , publisher =
- [76]
-
[77]
The Density Matrix Renormalization Group in Quantum Chemistry , year =
Chan, Garnet Kin-Lic and Sharma, Sandeep , journal =. The Density Matrix Renormalization Group in Quantum Chemistry , year =
-
[78]
Holmes, Adam A. and Tubman, Norm M. and Umrigar, C. J. , journal =. Heat-Bath Configuration Interaction: An Efficient Selected Configuration Interaction Algorithm Inspired by Heat-Bath Sampling , year =. doi:10.1021/acs.jctc.6b00407 , publisher =
-
[79]
Recent advances in multireference second order perturbation CI: The CIPSI method revisited , year =
Cimiraglia, Renzo and Persico, Maurizio , journal =. Recent advances in multireference second order perturbation CI: The CIPSI method revisited , year =
-
[80]
Booth, George H. and Thom, Alex J. W. and Alavi, Ali , journal =. 2009 , issn =
work page 2009
-
[81]
James J. Halson, Robert J. Anderson and George H. Booth , journal =. Improved stochastic multireference perturbation theory for correlated systems with large active spaces , year =. doi:10.1080/00268976.2020.1802072 , publisher =
-
[82]
Guther, Kai and Anderson, Robert J. and Blunt, Nick S. and Bogdanov, Nikolay A. and Cleland, Deidre and Dattani, Nike and Dobrautz, Werner and Ghanem, Khaldoon and Jeszenszki, Peter and Liebermann, Niklas and Manni, Giovanni Li and Lozovoi, Alexander Y. and Luo, Hongjun and Ma, Dongxia and Merz, Florian and Overy, Catherine and Rampp, Markus and Samanta, ...
work page 2020
-
[83]
Motta, Mario and Zhang, Shiwei , journal =. Ab initio computations of molecular systems by the auxiliary-field quantum Monte Carlo method , year =. doi:10.1002/wcms.1364 , keywords =
-
[84]
Lee, Joonho and Pham, Hung Q. and Reichman, David R. , journal =. Twenty Years of Auxiliary-Field Quantum Monte Carlo in Quantum Chemistry: An Overview and Assessment on Main Group Chemistry and Bond-Breaking , year =. doi:10.1021/acs.jctc.2c00802 , publisher =
-
[85]
Fermionic neural-network states for ab-initio electronic structure , year =
Choo, Kenny and Mezzacapo, Antonio and Carleo, Giuseppe , journal =. Fermionic neural-network states for ab-initio electronic structure , year =
-
[86]
Rath, Yannic and Booth, George H. , journal =. Framework for efficient ab initio electronic structure with Gaussian Process States , year =. doi:10.1103/PhysRevB.107.205119 , issue =
-
[87]
Journal of Chemical Theory and Computation , title =
Baiardi, Alberto and Kelemen, Anna Kl. Journal of Chemical Theory and Computation , title =. 2022 , issn =. doi:10.1021/acs.jctc.1c00984 , publisher =
-
[88]
Blunt, N. S. and Smart, Simon D. and Booth, George H. and Alavi, Ali , journal =. 2015 , issn =
work page 2015
-
[89]
and Opalka, Daniel and Overy, Catherine and Knowles, Peter J
Thomas, Robert E. and Opalka, Daniel and Overy, Catherine and Knowles, Peter J. and Alavi, Ali and Booth, George H. , journal =. 2015 , issn =
work page 2015
-
[90]
Chen, Siyuan and Zhang, Shiwei , journal =. Computation of forces and stresses in solids: Towards accurate structural optimization with auxiliary-field quantum Monte Carlo , year =. doi:10.1103/PhysRevB.107.195150 , issue =
-
[91]
Jiang, Tonghuan and Fang, Wei and Alavi, Ali and Chen, Ji , month =. General Analytical Nuclear Force and molecular potential energy surface from full configuration interaction quantum Monte Carlo , year =. 2204.13356 , eprinttype =
-
[92]
Ab Initio Molecular Dynamics with Quantum Monte Carlo , year =
Luo, Ye and Sorella, Sandro , journal =. Ab Initio Molecular Dynamics with Quantum Monte Carlo , year =
-
[93]
Lischka, Hans and Nachtigallov. Chemical Reviews , title =. 2018 , issn =. doi:10.1021/acs.chemrev.8b00244 , publisher =
-
[94]
Zur Quantentheorie der Molekeln , volume=. Annalen der Physik , author=. 1927 , month=. doi:10.1002/andp.19273892002 , number=
-
[95]
Nonadiabatic Events and Conical Intersections , year =
Matsika, Spiridoula and Krause, Pascal , journal =. Nonadiabatic Events and Conical Intersections , year =
-
[96]
Matsika, Spiridoula , journal =. Electronic Structure Methods for the Description of Nonadiabatic Effects and Conical Intersections , year =. doi:10.1021/acs.chemrev.1c00074 , publisher =
-
[97]
Ben-Nun, Michal and Martínez, Todd. J. , pages =. Ab Initio Quantum Molecular Dynamics , year =. Advances in Chemical Physics , doi =
-
[98]
Curchod, Basile F. E. and Mart. Chemical Reviews , title =. 2018 , issn =. doi:10.1021/acs.chemrev.7b00423 , publisher =
-
[99]
Nelson, Tammie R. and White, Alexander J. and Bjorgaard, Josiah A. and Sifain, Andrew E. and Zhang, Yu and Nebgen, Benjamin and Fernandez-Alberti, Sebastian and Mozyrsky, Dmitry and Roitberg, Adrian E. and Tretiak, Sergei , journal =. Non-adiabatic Excited-State Molecular Dynamics: Theory and Applications for Modeling Photophysics in Extended Molecular Ma...
-
[100]
Barbatti, Mario , journal =. Velocity Adjustment in Surface Hopping: Ethylene as a Case Study of the Maximum Error Caused by Direction Choice , year =. doi:10.1021/acs.jctc.1c00012 , publisher =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.