Recognition: 2 theorem links
· Lean TheoremG⁰W⁰ implementation based on the pseudopotential and numerical-atomic-orbital basis-set framework: Algorithms and benchmarks
Pith reviewed 2026-05-13 01:53 UTC · model grok-4.3
The pith
A G0W0 implementation within the numerical atomic orbital and pseudopotential framework reaches benchmark agreement with established codes through a compressed resolution of identity and analytic small-q handling.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that an NAO-PP G0W0 framework can be realized efficiently by combining the localized resolution of identity with a novel compression scheme, an analytic small-q treatment of the microscopic dielectric function, and real-space tensor filtering. These ingredients, together with a dielectric-function test for pseudopotential selection, produce quasiparticle band structures and gaps in excellent agreement with established G0W0 codes while delivering substantial computational gains.
What carries the argument
The localized resolution of identity (LRI) technique with a novel compression scheme, which reduces the cost of building the screened Coulomb interaction in the random phase approximation while preserving stability and accuracy.
If this is right
- G0W0 calculations become feasible for larger periodic systems within the NAO-PP basis than was previously practical.
- The analytic small-q treatment lowers the density of q-points required for converged dielectric functions.
- A dielectric-function inspection provides a concrete route to select appropriate pseudopotentials before performing G0W0.
- Real-space tensor filtering yields additional efficiency gains without degrading benchmark agreement.
Where Pith is reading between the lines
- The same compression and filtering steps could be reused in other many-body perturbation methods that rely on the random phase approximation.
- The NAO flexibility may simplify GW studies of low-dimensional or defective structures where plane-wave convergence is costly.
- Integration with machine-learning models trained on DFT data could supply corrected band gaps for dynamics or screening workflows.
Load-bearing premise
The compression scheme, real-space filtering, and analytic small-q treatment preserve the physical content of the dielectric response and self-energy without introducing errors larger than those tolerated in the benchmarks.
What would settle it
A calculation on silicon or a similar well-studied semiconductor in which the obtained direct or indirect band gap differs from accepted plane-wave G0W0 values by more than 0.1 eV.
Figures
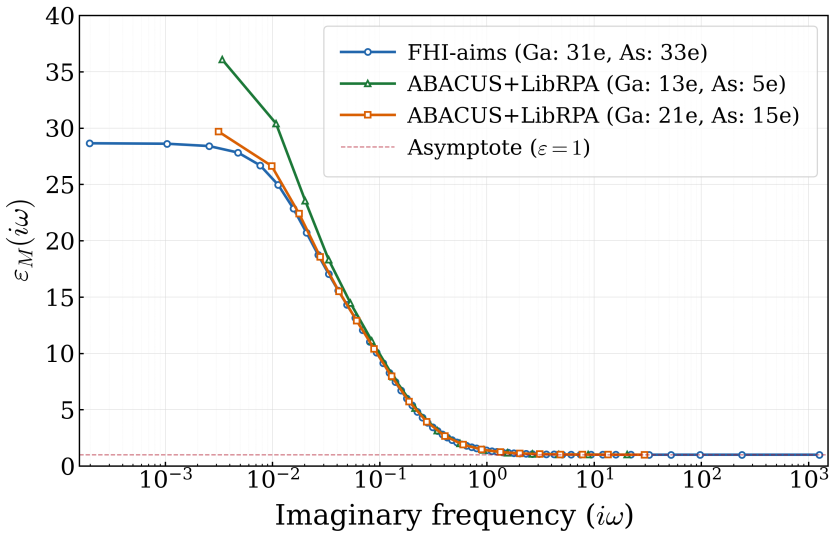
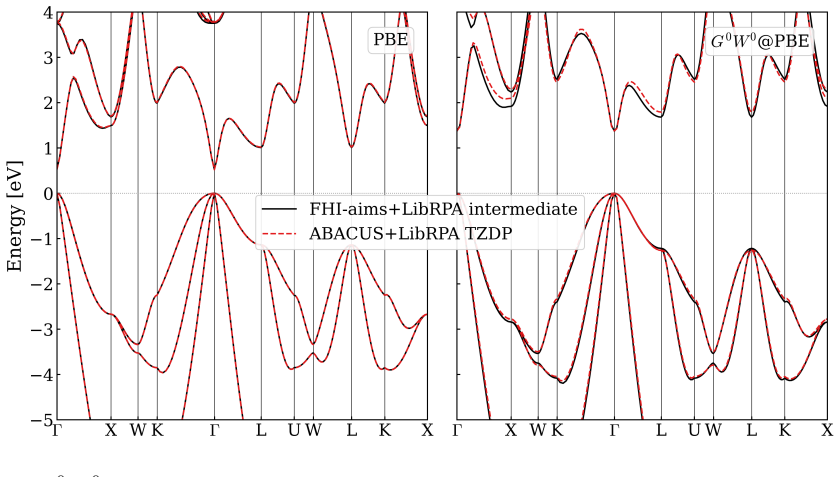



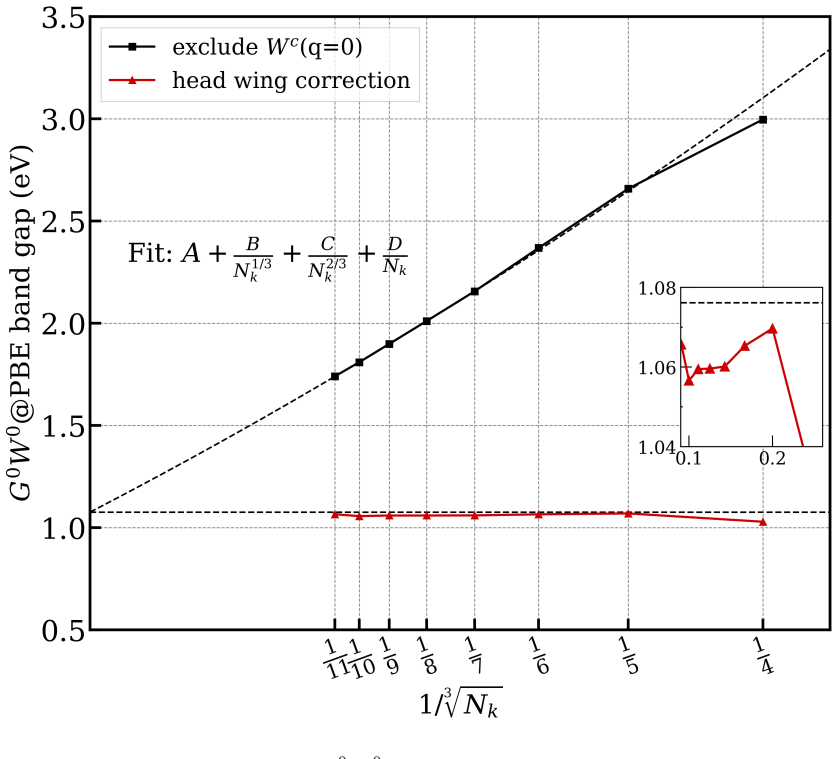





read the original abstract
The $GW$ method delivers substantially improved accuracy in electronic band structure calculations over conventional Kohn-Sham density functional theory (KS-DFT) by explicitly incorporating the electron self-energy effect beyond mean-field approximations. Despite many existing implementations, a periodic $GW$ implementation within the framework of numerical atomic orbitals (NAO) combined with the pseudopotential (PP) scheme has not been reported. This is urgently needed given the increasing popularity of the NAO-PP framework in KS-DFT calculations and its importance for the development of machine-learning electronic-structure approaches. In this work, we present an efficient NAO-PP-based $G^0W^0$ computational framework by interfacing the first-principles software package ABACUS with LibRPA -- a library for performing low-scaling random-phase approximation and $GW$ calculations based on NAOs. Our approach employs the localized resolution of identity (LRI) technique with a novel compression scheme, significantly improving both computational efficiency and numerical stability. In addition, an analytic treatment of the small-q limit of the microscopic dielectric function reduces the need for dense q-point sampling. Furthermore, we propose a practical strategy to select a suitable KS-DFT pseudopotential prior to $G^0W^0$ calculations by examining the frequency-dependent macroscopic dielectric function. Systematic benchmarks validate the effectiveness of our compression scheme and real-space tensor filtering strategies, demonstrating both high accuracy and significant computational efficiency gains. Comparisons with established $G^0W^0$ implementations show excellent agreement in band structures and band gaps, confirming ABACUS+LibRPA as a reliable and efficient platform for large-scale $G^0W^0$ simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents a G^0W^0 implementation within the ABACUS code using pseudopotentials and numerical atomic orbitals, interfaced with LibRPA. It introduces a novel LRI compression scheme for improved efficiency and stability, real-space tensor filtering, an analytic treatment of the small-q limit in the dielectric function, and a practical strategy for selecting KS-DFT pseudopotentials via the frequency-dependent macroscopic dielectric function. Systematic benchmarks on systems such as Si and GaAs demonstrate agreement with other established G^0W^0 codes and efficiency gains, supporting the claim that ABACUS+LibRPA is a reliable platform for large-scale G0W0 simulations.
Significance. If the accuracy of the new schemes holds, this fills a gap in periodic GW implementations for the increasingly popular NAO-PP framework and enables efficient large-system calculations relevant to materials science and machine-learning potentials. The benchmarks against other codes, focus on practical pseudopotential selection, and efficiency techniques are concrete strengths that could facilitate broader adoption.
major comments (2)
- [§3.2] §3.2: The LRI compression scheme is presented as key to efficiency and numerical stability, yet no systematic variation of the compression threshold is reported on supercells or molecules (where compression ratios are largest) to quantify effects on the dielectric matrix or self-energy. This is load-bearing for the large-scale reliability claim in the abstract.
- [real-space tensor filtering section] Real-space tensor filtering section: Benchmarks show good agreement on small primitive cells, but the manuscript provides no quantified error bounds or convergence tests with respect to filter cutoffs on larger systems where filtering is most active. Without these, it is unclear whether truncation errors remain controlled in the regime advertised for large-scale use.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and positive overall assessment of the work. We address each major comment point by point below and have revised the manuscript to incorporate additional validation tests as requested.
read point-by-point responses
-
Referee: [§3.2] §3.2: The LRI compression scheme is presented as key to efficiency and numerical stability, yet no systematic variation of the compression threshold is reported on supercells or molecules (where compression ratios are largest) to quantify effects on the dielectric matrix or self-energy. This is load-bearing for the large-scale reliability claim in the abstract.
Authors: We agree that explicit tests varying the LRI compression threshold on supercells and molecules would strengthen the validation of numerical stability and support the large-scale claims. In the revised manuscript we have added a dedicated subsection to §3.2 containing systematic threshold scans for a 2×2×2 Si supercell and for the benzene molecule. These new results quantify the impact on the dielectric matrix eigenvalues and on the quasiparticle self-energy corrections, confirming that errors remain below 5 meV for the threshold adopted in the production calculations. revision: yes
-
Referee: Real-space tensor filtering section: Benchmarks show good agreement on small primitive cells, but the manuscript provides no quantified error bounds or convergence tests with respect to filter cutoffs on larger systems where filtering is most active. Without these, it is unclear whether truncation errors remain controlled in the regime advertised for large-scale use.
Authors: We acknowledge that quantified error bounds with respect to the real-space filter cutoff on larger systems are needed to demonstrate control of truncation errors. In the revised manuscript we have extended the real-space tensor filtering section with convergence tests on Si and GaAs supercells (up to 64 atoms). A new table and accompanying discussion report the dependence of the dielectric matrix and self-energy on the cutoff, showing that the truncation error stays below 10 meV for the cutoffs used in the production runs. revision: yes
Circularity Check
No circularity: implementation and benchmarking paper with independent validation
full rationale
The manuscript describes an algorithmic implementation of G0W0 within the ABACUS+LibRPA framework using established GW equations, LRI compression, real-space filtering, and analytic small-q handling. These are presented as technical extensions rather than derivations. Benchmarks against external codes and standard test systems provide independent accuracy checks. No step reduces a claimed prediction or uniqueness result to a fitted parameter, self-citation chain, or redefinition of inputs. The reliability conclusion rests on numerical agreement with other implementations, not on internal consistency alone.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclearOur approach employs the localized resolution of identity (LRI) technique with a novel compression scheme... real-space tensor filtering strategies... analytic treatment of the small-q limit of the microscopic dielectric function
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanreality_from_one_distinction unclearSystematic benchmarks validate the effectiveness of our compression scheme and real-space tensor filtering strategies, demonstrating both high accuracy and significant computational efficiency gains.
Reference graph
Works this paper leans on
-
[1]
Compression scheme of LRI To demonstrate the effectiveness of the compression scheme proposed in Section IIIB, we perform G0W 0 calculations on MgO using both the conventional LRI scheme and the new compression scheme. A triple-zeta double-polarized (TZDP) basis set of the second generation developed for the ABACUS code[42] is employed for all elements, w...
-
[2]
Filtering of real-space tensors Due to the spatial locality of NAOs and the corresponding real-space representation of the quantities entering theG0W 0 workflow, the sparsity of the relevant atom-pair tensor blocks can be exploited to accelerate the computation. Following Ref.39, we apply a block-wise filtering algorithm to the dominant real-space tensors...
-
[3]
Convergence study Using Si as an example, we systematically examine the convergence behavior of the quasiparticle (QP) band gap with respect to five key computational parameters: (i) cutoff radius of basis functionsrcut, (ii)k-point sampling density, (iii) size of the auxiliary basis set, (iv) size of the NAO basis set, and (v) number of minimax points fo...
-
[4]
Benchmarking against plane-wave PA WG 0W 0 results (V ASP) To validate our implementation, we benchmark ourG0W 0 results against those obtained from the plane-wave projector-augmented-wave (PAW) method implemented in the VASP code. In the work of Klimeš et al.[8], G0W 0@PBE calculations were carried out using a plane-wave basis and norm-conserving PAW pot...
-
[5]
Benchmarking against NAO all-electronk-spaceG 0W 0 results (FHI-aims) Previous studies suggest that differentG0W 0 implementations still yield noticeable differ- ences in the calculated band gaps for certain materials [8, 13, 14, 16, 17, 68]. As previously alluded to, the differences may stem from different choices of basis sets, different treatments of v...
-
[6]
L. Hedin, New method for calculating the one-particle green’s function with application to the electron-gas problem, Phys. Rev.139, A796 (1965)
work page 1965
-
[7]
P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Physical Review136, B864 (1964), publisher: American Physical Society. 43
work page 1964
-
[8]
W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Physical Review140, A1133 (1965), publisher: American Physical Society
work page 1965
-
[9]
M. J. van Setten, F. Caruso, S. Sharifzadeh, X. Ren, M. Scheffler, F. Liu, J. Lischner, L. Lin, J. R. Deslippe, S. G. Louie, C. Yang, F. Weigend, J. B. Neaton, F. Evers, and P. Rinke, Gw100: Benchmarking g0w0 for molecular systems, Journal of Chemical Theory and Computation11, 5665 (2015), pMID: 26642984, https://doi.org/10.1021/acs.jctc.5b00453
-
[10]
D. Golze, M. Dvorak, and P. Rinke, The gw compendium: A practical guide to theoretical pho- toemission spectroscopy, Frontiers in ChemistryVolume 7 - 2019, 10.3389/fchem.2019.00377 (2019)
-
[11]
F. A. Rasmussen, P. S. Schmidt, K. T. Winther, and K. S. Thygesen, Efficient many-body calculations for two-dimensional materials using exact limits for the screened potential: Band gaps ofmos 2, h-bn, and phosphorene, Phys. Rev. B94, 155406 (2016)
work page 2016
-
[12]
L. Reining, The gw approximation: content, successes and limitations, WIREs Computational Molecular Science8, e1344 (2018)
work page 2018
-
[13]
J. c. v. Klimeš, M. Kaltak, and G. Kresse, Predictivegw calculations using plane waves and pseudopotentials, Phys. Rev. B90, 075125 (2014)
work page 2014
-
[14]
X. Gonze, G.-M. Rignanese, M. Verstraete, J.-M. Beuken, Y. Pouillon, R. Caracas, F. Jollet, M. Torrent, G. Zerah, M. Mikami, P. Ghosez, M. Veithen, J.-Y. Raty, V. Olevano, F. Bruneval, L. Reining, R. Godby, G. Onida, and D. H. D.C. Allan, A brief introduction to the ABINIT software package, Zeitschrift für Kristallographie - Crystalline Materials220, 558 (2005)
work page 2005
-
[15]
D. Sangalli, A. Ferretti, H. Miranda, C. Attaccalite, I. Marri, E. Cannuccia, P. Melo, M. Marsili, F. Paleari, A. Marrazzo, G. Prandini, P. Bonfa, M. Atambo, F. Affinito, M. Palummo, A. Molina- Sánchez, C. Hogan, M. Grüning, D. Varsano, and A. Marini, Many-body perturbation theory calculations using the yambo code, Journal of Physics: Condensed Matter31(2019)
work page 2019
-
[16]
J. Deslippe, G. Samsonidze, D. A. Strubbe, M. Jain, M. L. Cohen, and S. G. Louie, Berkeleygw: A massively parallel computer package for the calculation of the quasiparticle and optical properties of materials and nanostructures, Computer Physics Communications183, 1269 (2012)
work page 2012
- [17]
-
[18]
X. Ren, F. Merz, H. Jiang, Y. Yao, M. Rampp, H. Lederer, V. Blum, and M. Scheffler, 44 All-electron periodic G0W0 implementation with numerical atomic orbital basis functions: Algorithm and benchmarks, Phys. Rev. Mater.5, 013807 (2021)
work page 2021
- [19]
- [20]
-
[21]
T. Rangel, M. Del Ben, D. Varsano, G. Antonius, F. Bruneval, F. H. da Jornada, M. J. van Setten, O. K. Orhan, D. D. O’Regan, A. Canning, A. Ferretti, A. Marini, G.-M. Rignanese, J. Deslippe, S. G. Louie, and J. B. Neaton, Reproducibility ing0w0 calculations for solids, Comput. Phys. Commun.255, 107242 (2020)
work page 2020
- [22]
-
[23]
J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón, and D. Sánchez-Portal, The SIESTA method for ab initio order-N materials simulation, Journal of Physics: Condensed Matter14, 2745 (2002)
work page 2002
-
[24]
Ozaki, Variationally optimized atomic orbitals for large-scale electronic structures, Phys
T. Ozaki, Variationally optimized atomic orbitals for large-scale electronic structures, Phys. Rev. B67, 155108 (2003)
work page 2003
-
[25]
T. Ozaki and H. Kino, Numerical atomic basis orbitals from h to kr, Phys. Rev. B69, 195113 (2004)
work page 2004
-
[26]
P. Li, X. Liu, M. Chen, P. Lin, X. Ren, L. Lin, C. Yang, and L. He, Large-scale ab initio simulations based on systematically improvable atomic basis, Computational Materials Science 112, 503 (2016)
work page 2016
-
[27]
P. Lin, X. Ren, X. Liu, and L. He, Ab initio electronic structure calculations based on numerical atomic orbitals: Basic fomalisms and recent progresses, WIREs Computational Molecular Science14, e1687 (2024)
work page 2024
-
[28]
W. Zhou, D. Zheng, Q. Liu, D. Lu, Y. Liu, P. Lin, Y. Huang, X. Peng, J. J. Bao, C. Cai, Z. Jin, J. Wu, H. Zhang, G. Jin, Y. Ji, Z. Shen, X. Liu, L. Sun, Y. Cao, M. Sun, J. Liu, T. Chen, R. Liu, Y. Li, H. Han, X. Liang, T. Bao, Z. Deng, T. Liu, N. Chen, H. Ren, X. Zhang, Z. Liu, Y. Fu, M. Liu, Z. Li, T. Wen, Z. Tang, Y. Xu, W. Duan, X. Wang, Q. Gu, F.-Z. D...
work page 2025
-
[29]
H. Li, Z. Wang, N. Zou, M. Ye, R. Xu, X. Gong, W. Duan, and Y. Xu, Deep-learning density functional theory hamiltonian for efficient ab initio electronic-structure calculation, Nature Computational Science2, 367 (2022)
work page 2022
- [30]
-
[31]
Y. Wang, Y. Li, Z. Tang, H. Li, Z. Yuan, H. Tao, N. Zou, T. Bao, X. Liang, Z. Chen,et al., Universal materials model of deep-learning density functional theory hamiltonian, Science Bulletin69, 2514 (2024)
work page 2024
- [32]
- [33]
- [34]
- [35]
-
[36]
P. Lin, Y. Ji, L. He, X. Ren, and He, Efficient hybrid-functional-based force and stress calculations for periodic systems with thousands of atoms, J. Chem. Theory Comput. (2025), publisher: American Chemical Society
work page 2025
-
[37]
P. Lin, X. Ren, and L. He, Accuracy of Localized Resolution of the Identity in Periodic Hybrid Functional Calculations with Numerical Atomic Orbitals, The Journal of Physical Chemistry Letters11, 3082 (2020), publisher: American Chemical Society
work page 2020
-
[38]
P. Lin, X. Ren, and L. He, Efficient Hybrid Density Functional Calculations for Large Periodic Systems Using Numerical Atomic Orbitals, Journal of Chemical Theory and Computation17, 222 (2021)
work page 2021
-
[39]
Y. Cao, M.-Y. Zhang, P. Lin, M. Chen, and X. Ren, Applying space-group symmetry 46 to speed up hybrid-functional calculations within the framework of numerical atomic or- bitals, Journal of Chemical Theory and Computation21, 8086 (2025), pMID: 40789044, https://doi.org/10.1021/acs.jctc.5c00537
-
[40]
S. V. Levchenko, X. Ren, J. Wieferink, R. Johanni, P. Rinke, V. Blum, and M. Scheffler, Hybrid functionals for large periodic systems in an all-electron, numeric atom-centered basis framework, Computer Physics Communications192, 60 (2015)
work page 2015
- [41]
-
[42]
R. Shi, P. Lin, M.-Y. Zhang, L. He, and X. Ren, Subquadratic-scaling real-space random phase approximation correlation energy calculations for periodic systems with numerical atomic orbitals, Phys. Rev. B109, 035103 (2024)
work page 2024
- [43]
-
[44]
M.-Y. Zhang, P. Lin, R. Shi, and X. Ren, Low-scalinggw calculation of quasi-particle energies within numerical atomic orbital framework (2026), arXiv:2603.27292
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[45]
M. Chen, G.-C. Guo, and L. He, Systematically improvable optimized atomic basis sets forab initiocalculations, Journal of Physics: Condensed Matter22, 445501 (2010)
work page 2010
-
[46]
M. Chen, G.-C. Guo, and L. He, Electronic structure interpolation via atomic orbitals, Journal of Physics: Condensed Matter23, 325501 (2011)
work page 2011
-
[47]
P. Lin, X. Ren, and L. He, Strategy for constructing compact numerical atomic orbital basis sets by incorporating the gradients of reference wavefunctions, Physical Review B103, 235131 (2021)
work page 2021
-
[48]
M. van Setten, M. Giantomassi, E. Bousquet, M. Verstraete, D. Hamann, X. Gonze, and G.-M. Rignanese, The pseudodojo: Training and grading a 85 element optimized norm-conserving pseudopotential table, Computer Physics Communications226, 39 (2018)
work page 2018
-
[49]
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter, and M. Scheffler,Ab initio molecular simulations with numeric atom-centered orbitals, Computer Physics Communications 180, 2175 (2009)
work page 2009
-
[50]
J. Hafner, Ab-initio simulations of materials using VASP: Density-functional theory and beyond, 47 Journal of Computational Chemistry29, 2044 (2008)
work page 2044
-
[51]
H. N. Rojas, R. W. Godby, and R. J. Needs, Space-time method for ab initio calculations of self-energies and dielectric response functions of solids, Phys. Rev. Lett.74, 1827 (1995)
work page 1995
-
[52]
M. M. Rieger, L. Steinbeck, I. White, H. Rojas, and R. Godby, The gw space-time method for the self-energy of large systems, Computer Physics Communications117, 211 (1999)
work page 1999
- [53]
- [54]
-
[55]
P. Liu, M. Kaltak, J. c. v. Klimeš, and G. Kresse, Cubic scalinggw: Towards fast quasiparticle calculations, Phys. Rev. B94, 165109 (2016)
work page 2016
-
[56]
M. Azizi, J. Wilhelm, D. Golze, M. Giantomassi, R. L. Panadés-Barrueta, F. A. Delesma, A. Buccheri, A. Gulans, P. Rinke, C. Draxl, and X. Gonze, Time-frequency component of the greenx library: minimax grids for efficient rpa and gw calculations, Journal of Open Source Software8, 5570 (2023)
work page 2023
- [57]
-
[58]
X.-Z. Li, R. Gómez-Abal, H. Jiang, C. Ambrosch-Draxl, and M. Scheffler, Impact of widely used approximations to the g0w0 method: an all-electron perspective, New Journal of Physics 14, 023006 (2012)
work page 2012
-
[59]
R. Gómez-Abal, X. Li, M. Scheffler, and C. Ambrosch-Draxl, Influence of the core-valence inter- action and of the pseudopotential approximation on the electron self-energy in semiconductors, Phys. Rev. Lett.101, 106404 (2008)
work page 2008
-
[60]
A. C. Ihrig, J. Wieferink, I. Y. Zhang, M. Ropo, X. Ren, P. Rinke, M. Scheffler, and V. Blum, Accurate localized resolution of identity approach for linear-scaling hybrid density functionals and for many-body perturbation theory, New Journal of Physics17, 093020 (2015), publisher: IOP Publishing
work page 2015
-
[61]
W. P. Huhn and V. Blum, One-hundred-three compound band-structure benchmark of post- self-consistent spin-orbit coupling treatments in density functional theory, Phys. Rev. Mater.1, 48 033803 (2017)
work page 2017
-
[62]
D. R. Hamann, Optimized norm-conserving vanderbilt pseudopotentials, Phys. Rev. B88, 085117 (2013)
work page 2013
-
[63]
J. Spencer and A. Alavi, Efficient calculation of the exact exchange energy in periodic systems using a truncated coulomb potential, Phys. Rev. B77, 193110 (2008)
work page 2008
-
[64]
C. Friedrich, A. Schindlmayr, and S. Blügel, Efficient calculation of the coulomb matrix and its expansion around k=0 within the flapw method, Computer Physics Communications180, 347 (2009)
work page 2009
-
[65]
C. Friedrich, S. Blügel, and A. Schindlmayr, Efficient implementation of thegw approximation within the all-electron flapw method, Phys. Rev. B81, 125102 (2010)
work page 2010
- [66]
-
[67]
G. Jin, H. Pang, Y. Ji, Z. Dai, and L. He, Pyatb: An efficient python package for electronic structure calculations using ab initio tight-binding model, Computer Physics Communications 291, 108844 (2023)
work page 2023
- [68]
-
[69]
C. Freysoldt, P. Eggert, P. Rinke, A. Schindlmayr, R. Godby, and M. Scheffler, Dielectric anisotropy in the gw space–time method, Computer Physics Communications176, 1 (2007)
work page 2007
-
[70]
X. Ren, P. Rinke, V. Blum, J. Wieferink, A. Tkatchenko, A. Sanfilippo, K. Reuter, and M. Scheffler, Resolution-of-identity approach to Hartree–Fock, hybrid density functionals, RPA, MP2 and GW with numeric atom-centered orbital basis functions, New Journal of Physics14, 053020 (2012), publisher: IOP Publishing
work page 2012
-
[71]
A. Goncharsky, V. Stepanov, A. Tikhonov, and A. Yagola, Numerical Methods for the Solution of Ill-Posed Problems(Kluwer Academic Publishers, Dordrecht, 1995)
work page 1995
-
[72]
M. Shishkin and G. Kresse, Implementation and performance of the frequency-dependentgw method within the paw framework, Phys. Rev. B74, 035101 (2006)
work page 2006
-
[73]
H. Jiang and P. Blaha,gw with linearized augmented plane waves extended by high-energy 49 local orbitals, Phys. Rev. B93, 115203 (2016)
work page 2016
-
[74]
I. Vurgaftman, J. R. Meyer, and L. R. Ram-Mohan, Band parameters for iii–v compound semiconductors and their alloys, J. Appl. Phys.89, 5815 (2001). 50
work page 2001
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.