Recognition: 2 theorem links
· Lean TheoremHessian Matching for Machine-Learned Coarse-Grained Molecular Dynamics
Pith reviewed 2026-05-14 19:50 UTC · model grok-4.3
The pith
Adding stochastic Hessian-vector product matching to force matching improves coarse-grained molecular dynamics models by incorporating curvature information.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Coarse-grained (CG) molecular dynamics enables simulations at inaccessible timescales, but existing neural potentials trained via force matching capture only the gradient of the free-energy surface. We introduce stochastic Hessian-vector product (HVP) matching that augments force matching with second-order curvature information without constructing the full Hessian. A decomposition separates the target CG Hessian into a model-independent projected all-atom Hessian precomputed once and a model-dependent covariance correction computed online. An unbiased estimator uses random probe vectors for the matching objective. On nine fast-folding proteins, HVP matching outperforms force matching on 8,
What carries the argument
The decomposition of the target CG Hessian into a model-independent projected AA Hessian and a model-dependent covariance correction, combined with random probe vectors for unbiased stochastic estimation of the Hessian-matching objective.
If this is right
- CG potentials now constrain both the gradient and the curvature of the underlying free-energy surface.
- Slow-mode metrics improve, with up to 85% lower KL divergence on the largest test protein.
- The method remains computationally practical because the AA Hessian projection is precomputed and the correction uses cheap online covariance estimates.
- Better reproduction of collective motions leads to more accurate long-timescale biomolecular dynamics.
- The approach demonstrates that higher-order physical information can be instilled into learned potentials without prohibitive cost.
Where Pith is reading between the lines
- Extending the Hessian matching to other coarse-graining schemes or non-protein systems could reveal where curvature information matters most.
- If the unbiased estimator property holds more generally, similar techniques might apply to machine-learned potentials in other domains like materials science.
- The improvements in slow modes suggest that such models could enable more reliable exploration of conformational changes in drug design or protein folding studies.
- One could test whether adding even higher-order terms like third derivatives further enhances transferability.
Load-bearing premise
The decomposition of the target CG Hessian into a model-independent projected AA Hessian plus a model-dependent covariance correction remains valid and the stochastic HVP estimator stays unbiased throughout training.
What would settle it
A direct comparison on the nine-protein benchmark where HVP matching fails to reduce the Kullback-Leibler divergence on slow modes relative to force matching would falsify the performance claim.
Figures

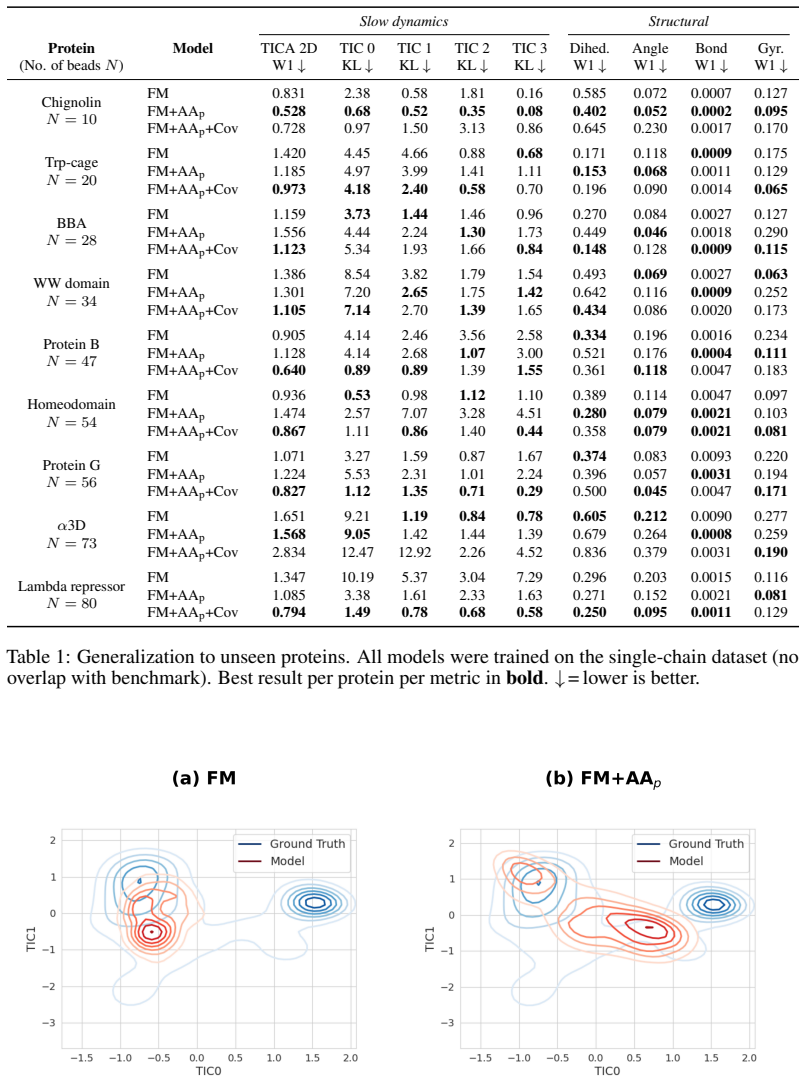

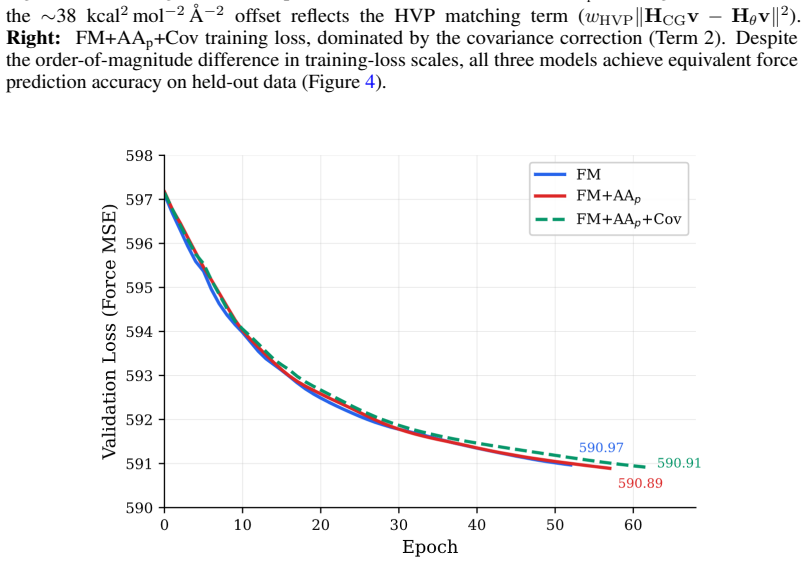
read the original abstract
Coarse-grained (CG) molecular dynamics enables simulations of atomic systems such as biomolecules at timescales inaccessible to all-atom (AA) methods, but existing CG neural potentials trained via force matching capture only the gradient of the free-energy surface, leaving its curvature unconstrained. We introduce a framework that augments force matching with stochastic Hessian-vector product (HVP) matching, instilling second-order curvature information into CG potentials without constructing the full Hessian. We derive a decomposition of the target CG Hessian into a model-independent projected AA Hessian, precomputed once before training, and a model-dependent covariance correction computed online at negligible cost. We construct an unbiased stochastic estimator of the Hessian-matching objective by using random probe vectors. We evaluate our method by comparing against force matching on a benchmark of nine fast-folding proteins unseen during training. HVP matching outperforms plain force matching on 8 of 9 proteins on slow-mode metrics, with reductions of up to 85% in the Kullback--Leibler divergence between the CG and reference distributions along the slowest collective mode of the largest protein. Our results demonstrate that higher-order physical supervision is a practical path to more accurate and transferable CG potentials for biomolecular simulation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents Hessian Matching, which augments standard force matching with stochastic Hessian-vector product (HVP) matching to incorporate second-order curvature information into machine-learned coarse-grained (CG) molecular dynamics potentials. The key technical contribution is a derived decomposition of the target CG Hessian into a model-independent projected all-atom Hessian (precomputed once) and a model-dependent covariance correction (computed online), allowing construction of an unbiased stochastic estimator via random probe vectors without forming the full Hessian. Empirical evaluation on nine fast-folding proteins shows that the method outperforms plain force matching on slow-mode metrics for eight proteins, achieving up to 85% reduction in Kullback-Leibler divergence along the slowest collective mode for the largest system.
Significance. If the derivation is rigorous and the estimator unbiased, this provides an efficient path to higher-order supervision in CG potential training, potentially yielding models that better capture the free-energy surface curvature and thus improve accuracy in long-timescale biomolecular simulations. The reported gains on slow-mode distributions are notable and suggest practical benefits for transferability across proteins unseen in training.
major comments (2)
- [§3.2] §3.2 (decomposition): The claim that the target CG Hessian decomposes exactly into a precomputed projected AA Hessian plus an online covariance correction must be shown to remain exact and yield an unbiased stochastic HVP estimator when the covariance term is recomputed from the current model parameters at each training step; finite-sample effects or implicit assumptions in the projection could otherwise make the added loss match an incorrect curvature.
- [§4.3] §4.3 (benchmark results): The reported outperformance on 8 of 9 proteins and the 85% KL reduction for the largest protein rest on the slow-mode metrics; the paper should include controls confirming that these gains are not attributable to implicit regularization from the online covariance term rather than faithful second-order supervision.
minor comments (2)
- [§2.1] §2.1: The notation distinguishing the AA-to-CG mapping operator from the Hessian projection could be made more explicit with a short summary equation or diagram.
- [Figure 4] Figure 4 caption: Add the number of independent runs and any statistical significance tests supporting the per-protein comparisons.
Simulated Author's Rebuttal
We thank the referee for their insightful and constructive comments on our manuscript. We provide point-by-point responses to the major comments below, indicating where revisions have been made to strengthen the paper.
read point-by-point responses
-
Referee: [§3.2] §3.2 (decomposition): The claim that the target CG Hessian decomposes exactly into a precomputed projected AA Hessian plus an online covariance correction must be shown to remain exact and yield an unbiased stochastic HVP estimator when the covariance term is recomputed from the current model parameters at each training step; finite-sample effects or implicit assumptions in the projection could otherwise make the added loss match an incorrect curvature.
Authors: The decomposition follows directly from the definition of the coarse-grained potential of mean force obtained by marginalizing the all-atom Boltzmann distribution. The projected AA Hessian term is independent of the CG model parameters and is precomputed exactly once from the reference ensemble. The covariance correction is the exact conditional variance term required by the marginalization identity and is therefore the correct target Hessian for whatever CG parameters are present at a given training step. Recomputing it online preserves exactness. The stochastic estimator remains unbiased because the expectation of the random-probe outer product recovers the matrix-vector product for each term separately; this linearity holds regardless of parameter values. Finite-sample effects are controlled by the number of probes (10 in our experiments), and the projection is exact under the linear coarse-graining map employed. We have added a clarifying paragraph in the revised §3.2 and placed the full algebraic derivation in the appendix. revision: partial
-
Referee: [§4.3] §4.3 (benchmark results): The reported outperformance on 8 of 9 proteins and the 85% KL reduction for the largest protein rest on the slow-mode metrics; the paper should include controls confirming that these gains are not attributable to implicit regularization from the online covariance term rather than faithful second-order supervision.
Authors: We agree that an explicit control is useful to isolate the source of the improvement. In the revised manuscript we have added an ablation in §4.3 that trains three variants on the same data: (i) standard force matching, (ii) force matching augmented only by the online covariance correction (no HVP loss), and (iii) the full Hessian-matching objective. The covariance-only variant yields only marginal gains on slow-mode KL divergence, whereas the complete second-order objective reproduces the reported 8-of-9 outperformance and the 85% reduction on the largest system. These new results appear in the updated Figure 5 and accompanying text, confirming that the observed benefits arise from faithful curvature supervision rather than incidental regularization. revision: yes
Circularity Check
No significant circularity in derivation or objective
full rationale
The paper derives a decomposition of the target CG Hessian into a precomputed model-independent projected AA Hessian plus an online model-dependent covariance correction, then constructs an unbiased stochastic HVP estimator using random probes. The central performance claims rest on empirical comparison against force matching on nine unseen proteins, with metrics such as KL divergence on slow modes. No equation reduces by construction to a fitted parameter defined in terms of the model output itself, no load-bearing self-citation chain is invoked to justify uniqueness or the ansatz, and the training objective is not statistically forced to match its own inputs. The method remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The target coarse-grained Hessian decomposes into a model-independent projected all-atom Hessian and a model-dependent covariance correction term.
- standard math Random probe vectors yield an unbiased estimator of the Hessian-matching objective.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
HCG = ⟨ΞF HAA ΞTF⟩R − β Σ(ΞF FAA, ΞF FAA) (Eq. 9); stochastic HVP matching loss LHVP with random probes vk
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
decomposition of target CG Hessian into model-independent projected AA Hessian plus model-dependent covariance correction
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Shaw et al
David E. Shaw et al. Millisecond-scale molecular dynamics simulations on anton. InProceed- ings of the Conference on High Performance Computing Networking, Storage and Analysis, SC ’09, page 1–11. ACM, November 2009
2009
-
[2]
Shaw et al
David E. Shaw et al. Anton 3: Twenty microseconds of molecular dynamics simulation before lunch. InSC21: International Conference for High Performance Computing, Networking, Storage and Analysis, pages 1–11, 2021
2021
-
[3]
W. G. Noid. Perspective: Coarse-grained models for biomolecular systems.The Journal of Chemical Physics, 139(9), September 2013
2013
-
[4]
Coarse-grained protein models and their applications.Chemical Reviews, 116(14):7898–7936, June 2016
Sebastian Kmiecik, Dominik Gront, Michal Kolinski, Lukasz Wieteska, Aleksandra Elzbieta Dawid, and Andrzej Kolinski. Coarse-grained protein models and their applications.Chemical Reviews, 116(14):7898–7936, June 2016
2016
-
[5]
Ercolessi and J
F. Ercolessi and J. B. Adams. Interatomic potentials from first-principles calculations: The force-matching method.Europhysics Letters, 26(8):583–588, 1994
1994
-
[6]
Sergei Izvekov and Gregory A. V oth. A multiscale coarse-graining method for biomolecular systems.The Journal of Physical Chemistry B, 109(7):2469–2473, 2005
2005
-
[7]
W. G. Noid, Jhih-Wei Chu, Gary S. Ayton, Vinod Krishna, Sergei Izvekov, Gregory A. V oth, Avisek Das, and Hans C. Andersen. The multiscale coarse-graining method. I. A rigorous bridge between atomistic and coarse-grained models.The Journal of Chemical Physics, 128(24):244114, 2008
2008
-
[8]
Machine learning of coarse-grained molecular dynamics force fields.ACS Central Science, 5(5):755–767, 2019
Jiang Wang, Simon Olsson, Christoph Wehmeyer, Adrià Pérez, Nicholas E Charron, Gianni de Fabritiis, Frank Noé, and Cecilia Clementi. Machine learning of coarse-grained molecular dynamics force fields.ACS Central Science, 5(5):755–767, 2019
2019
-
[9]
Coarse graining molecular dynamics with graph neural networks.The Journal of Chemical Physics, 153(19):194101, 2020
Brooke E Husic, Nicholas E Charron, Dominik Lemm, Jiang Wang, Adrià Pérez, Maciej Majewski, Andreas Krämer, Yaoyi Chen, Simon Olsson, Gianni de Fabritiis, Frank Noé, and Cecilia Clementi. Coarse graining molecular dynamics with graph neural networks.The Journal of Chemical Physics, 153(19):194101, 2020
2020
-
[10]
Deep coarse-grained potentials via relative entropy minimization.The Journal of Chemical Physics, 157(24):244103, December 2022
Stephan Thaler, Maximilian Stupp, and Julija Zavadlav. Deep coarse-grained potentials via relative entropy minimization.The Journal of Chemical Physics, 157(24):244103, December 2022
2022
-
[11]
Flow-matching: Efficient coarse-graining of molecular dynamics without forces.Journal of Chemical Theory and Computation, 19(3):942–952, February 2023
Jonas Köhler, Yaoyi Chen, Andreas Krämer, Cecilia Clementi, and Frank Noé. Flow-matching: Efficient coarse-graining of molecular dynamics without forces.Journal of Chemical Theory and Computation, 19(3):942–952, February 2023
2023
-
[12]
Charron, Toni Giorgino, Brooke E
Maciej Majewski, Adrià Pérez, Philipp Thölke, Stefan Doerr, Nicholas E. Charron, Toni Giorgino, Brooke E. Husic, Cecilia Clementi, Frank Noé, and Gianni De Fabritiis. Ma- chine learning coarse-grained potentials of protein thermodynamics.Nature Communications, 14(1):5739, 2023
2023
-
[13]
Beyond numerical Hessians: Higher-order derivatives for machine learning in- teratomic potentials via automatic differentiation.Journal of Chemical Theory and Computation, 2024
Niklas Fang et al. Beyond numerical Hessians: Higher-order derivatives for machine learning in- teratomic potentials via automatic differentiation.Journal of Chemical Theory and Computation, 2024
2024
-
[14]
Yuan, Anup Kumar, Xingyi Guan, Eric D
Eric C.-Y . Yuan, Anup Kumar, Xingyi Guan, Eric D. Hermes, Andrew S. Rosen, Judit Zádor, Teresa Head-Gordon, and Samuel M. Blau. Analytical ab initio hessian from a deep learning potential for transition state optimization.Nature Communications, 2024
2024
-
[15]
Smith, and Jose L
Austin Rodriguez, Justin S. Smith, and Jose L. Mendoza-Cortes. Does Hessian data improve the performance of machine learning potentials?Journal of Chemical Theory and Computation, 2025
2025
-
[16]
Austin Rodriguez, Justin S. Smith, and Jose L. Mendoza-Cortes. Projected Hessian learning: Fast curvature supervision for accurate machine-learning interatomic potentials.arXiv preprint arXiv:2603.04523, 2026. 10
-
[17]
Shoot from the HIP: Hessian interatomic potentials without derivatives, 2025
Andreas Burger, Luca Thiede, Nikolaj Rønne, Varinia Bernales, Nandita Vijaykumar, Tejs Vegge, Arghya Bhowmik, and Alan Aspuru-Guzik. Shoot from the HIP: Hessian interatomic potentials without derivatives, 2025
2025
-
[18]
Charron et al
Nicholas E. Charron et al. Navigating protein landscapes with a machine-learned transferable coarse-grained model.Nature Chemistry, 17(8):1284–1292, 2025
2025
-
[19]
Blue moon sampling, vectorial reaction coordinates, and unbiased constrained dynamics.ChemPhysChem, 6(9):1809–1814, 2005
Giovanni Ciccotti, Raymond Kapral, and Eric Vanden-Eijnden. Blue moon sampling, vectorial reaction coordinates, and unbiased constrained dynamics.ChemPhysChem, 6(9):1809–1814, 2005
2005
-
[20]
Katsoulakis, and Petr Plechá ˇc
Evangelia Kalligiannaki, Vagelis Harmandaris, Markos A. Katsoulakis, and Petr Plechá ˇc. The geometry of generalized force matching and related information metrics in coarse-graining of molecular systems.The Journal of Chemical Physics, 143(8):084105, 2015
2015
-
[21]
Deep learning via Hessian-free optimization
James Martens. Deep learning via Hessian-free optimization. InInternational Conference on Machine Learning, pages 735–742, 2010
2010
-
[22]
Optimizing neural networks with Kronecker-factored approximate curvature
James Martens and Roger Grosse. Optimizing neural networks with Kronecker-factored approximate curvature. InInternational Conference on Machine Learning, pages 2408–2417, 2015
2015
-
[23]
Pearlmutter
Barak A. Pearlmutter. Fast exact multiplication by the Hessian.Neural Computation, 6(1):147– 160, 1994
1994
-
[24]
Carter, Giovanni Ciccotti, James T
E.A. Carter, Giovanni Ciccotti, James T. Hynes, and Raymond Kapral. Constrained reaction co- ordinate dynamics for the simulation of rare events.Chemical Physics Letters, 156(5):472–477, April 1989
1989
-
[25]
Kidder, Ryan J
Katherine M. Kidder, Ryan J. Szukalo, and W. G. Noid. Energetic and entropic considerations for coarse-graining.The European Physical Journal B, 94(7), July 2021
2021
-
[26]
Maier, Carmenza Martinez, Koushik Kasavajhala, Lauren Wickstrom, Kevin E
James A. Maier, Carmenza Martinez, Koushik Kasavajhala, Lauren Wickstrom, Kevin E. Hauser, and Carlos Simmerling. ff14sb: Improving the accuracy of protein side chain and backbone parameters from ff99sb.Journal of Chemical Theory and Computation, 11(8):3696–3713, July 2015
2015
-
[27]
Openmm 7: Rapid development of high performance algorithms for molecular dynamics.PLOS Computational Biology, 13(7):e1005659, July 2017
Peter Eastman et al. Openmm 7: Rapid development of high performance algorithms for molecular dynamics.PLOS Computational Biology, 13(7):e1005659, July 2017
2017
-
[28]
A standardized benchmark for machine- learned molecular dynamics using weighted ensemble sampling.The Journal of Physical Chemistry B, 129(50):12828–12840, December 2025
Alexander Aghili, Andy Bruce, Daniel Sabo, Sanya Murdeshwar, Kevin Bachelor, Ionut Mistreanu, Ashwin Lokapally, and Razvan Marinescu. A standardized benchmark for machine- learned molecular dynamics using weighted ensemble sampling.The Journal of Physical Chemistry B, 129(50):12828–12840, December 2025
2025
-
[29]
TorchMD: A deep learning framework for molecular simulations.Journal of Chemical Theory and Computation, 17(4):2355–2363, 2021
Stefan Doerr, Maciej Majewski, Adrià Pérez, Andreas Krämer, Cecilia Clementi, Frank Noe, Toni Giorgino, and Gianni De Fabritiis. TorchMD: A deep learning framework for molecular simulations.Journal of Chemical Theory and Computation, 17(4):2355–2363, 2021
2021
-
[30]
Pelaez, Guillem Simeon, Raimondas Galvelis, Antonio Mirarchi, Peter Eastman, Stefan Doerr, Philipp Thölke, Thomas E
Raul P. Pelaez, Guillem Simeon, Raimondas Galvelis, Antonio Mirarchi, Peter Eastman, Stefan Doerr, Philipp Thölke, Thomas E. Markland, and Gianni De Fabritiis. Torchmd-net 2.0: Fast neural network potentials for molecular simulations, 2024
2024
-
[31]
Decoupled weight decay regularization
Ilya Loshchilov and Frank Hutter. Decoupled weight decay regularization. InInternational Conference on Learning Representations, 2019
2019
-
[32]
Identification of slow molecular order parameters for markov model construction.The Journal of Chemical Physics, 139(1), July 2013
Guillermo Pérez-Hernández, Fabian Paul, Toni Giorgino, Gianni De Fabritiis, and Frank Noé. Identification of slow molecular order parameters for markov model construction.The Journal of Chemical Physics, 139(1), July 2013. 11 A The Hessian for nonlinear coarse graining In Section 2 of the main paper, we derived the Hessian for linear coarse-graining maps....
2013
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.