BioXArena: Benchmarking LLM Agents on Multi-Modal Biomedical Machine Learning Tasks
Pith reviewed 2026-05-19 19:27 UTC · model grok-4.3
The pith
BioXArena tests whether LLM agents can write code to build predictive models across 76 multi-modal biomedical tasks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
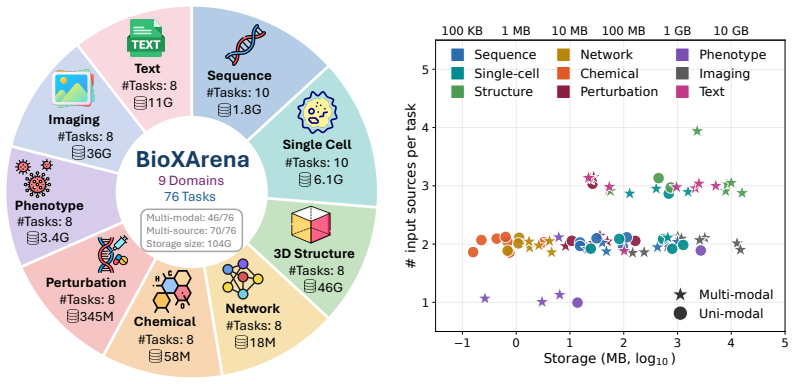
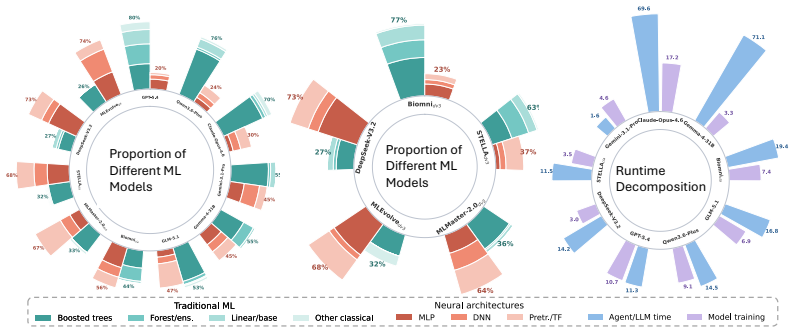
BioXArena contains 76 end-to-end tasks across nine domains that require agents to generate executable code, train predictive models on multi-modal inputs such as images, sequences, and omics matrices, and submit predictions against hidden labels using normalized biology-aware metrics. When eleven agent configurations are tested in a standardized environment, MLEvolve with Gemini-3.1-Pro records the highest average score of 0.666 and GPT-5.4 follows at 0.636, with performance varying substantially by domain, model backbone, and agent scaffold.
What carries the argument
BioXArena, the unified evaluation framework that curates tasks from primary sources, supplies hidden test labels, and applies biology-aware metrics to score agent-generated code and model submissions.
If this is right
- Agent performance depends on the choice of LLM backbone and scaffold rather than a single configuration working across all domains.
- Ablation and scaling studies can isolate how inference budget, cost, and domain characteristics affect coding success.
- The benchmark supplies standardized runners and graders that allow direct comparison of future agent designs on the same tasks.
- Failure-mode analysis reveals where current agents struggle with multi-modal integration or biology-specific constraints.
Where Pith is reading between the lines
- The moderate top scores suggest that further gains may require agents that better combine outputs across different data modalities.
- Domain variation implies that specialized biomedical knowledge or retrieval could narrow performance gaps between domains.
- Public release of tasks, graders, and trajectories creates a shared testbed for tracking progress in automated scientific coding.
Load-bearing premise
The 76 curated tasks with hidden labels and biology-aware metrics accurately and fairly measure real-world agent performance on heterogeneous multi-modal biomedical ML problems.
What would settle it
High-scoring agents on BioXArena produce models that fail to generalize or yield poor predictions when tested on new, independent biomedical datasets drawn from the same domains but not present in the benchmark.
Figures





read the original abstract
Large language model (LLM) agents are increasingly capable of automating components of machine learning development, yet existing biomedical benchmarks mainly focus on question answering, reasoning, and tool usage, or evaluate only narrow aspects of biomedical ML coding. We present BioXArena, a biomedical machine learning benchmark designed to evaluate whether agents can generate task-specific model training pipelines for heterogeneous and multi-modal biomedical datasets. BioXArena contains 76 end-to-end tasks across 9 domains, including sequence modeling, single-cell analysis, structural biology, network biology, chemical biology, perturbation dynamics, phenotype-disease modeling, biomedical imaging, and text-integrated learning. Each task is curated from primary biomedical sources into a unified evaluation framework with hidden labels, held-out graders, and biology-aware metrics normalized to a 0 to 1 scale. Agents are required to write executable code, train predictive models, and generate submissions for private test samples. Most tasks involve multiple input modalities, including tabular data, images, natural language, molecular sequences, omics matrices, and protein structures. We evaluate 11 agent configurations in a standardized 2-hour single-GPU environment. MLEvolve with Gemini-3.1-Pro achieves the highest average score of 0.666, followed by GPT-5.4 with 0.636, while no single agent consistently dominates across all domains. We additionally perform extensive ablation studies, robustness evaluations, scaling analyses, cost analyses, and failure-mode investigations to better understand how model backbones, agent scaffolds, inference budgets, and biomedical domains influence BioML coding performance. We will publicly release all benchmark tasks, graders, execution runners, leaderboard results, and agent trajectories.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces BioXArena, a benchmark consisting of 76 end-to-end tasks across 9 biomedical ML domains (sequence modeling, single-cell analysis, structural biology, etc.). LLM agents must write executable code to train predictive models on multi-modal inputs and submit predictions for private test sets with hidden labels. Biology-aware metrics are normalized to [0,1]; 11 agent configurations are evaluated in a standardized 2-hour single-GPU setting. MLEvolve with Gemini-3.1-Pro achieves the highest average score of 0.666, followed by GPT-5.4 at 0.636, with no agent dominating all domains. The paper includes ablation studies, robustness checks, scaling analyses, cost analyses, and failure-mode investigations, and commits to public release of tasks, graders, runners, and trajectories.
Significance. If the tasks and metrics prove robust, BioXArena would address a clear gap by evaluating full ML pipeline generation rather than isolated QA or narrow coding on biomedical data. The standardized single-GPU environment, extensive ablations across backbones/scaffolds/domains, and public release of all components (including agent trajectories) are concrete strengths that support reproducibility and community extensions. The multi-modal coverage and biology-aware metrics, if properly calibrated, could yield actionable insights into where current agents succeed or fail on realistic biomedical problems.
major comments (2)
- [Abstract and §4] Abstract and §4 (Metrics and Evaluation): The headline average scores (0.666 and 0.636) and the claim that 'no single agent consistently dominates across all domains' rest on cross-domain comparability of biology-aware metrics normalized to [0,1]. The manuscript provides no explicit description of the normalization procedure, shared statistical grounding, expert baselines, or handling of differing difficulty floors across domains (e.g., AUC thresholds in imaging versus stricter biology-specific criteria in omics). If normalization is performed independently per task without calibration, domain scores become incomparable and the reported averages lose interpretability.
- [§3] §3 (Task Curation and Validation): The curation of 76 tasks from primary sources into a unified framework with hidden labels and held-out graders is load-bearing for the central claim that the benchmark 'accurately and fairly measure[s] real-world agent performance.' The manuscript states the design but supplies insufficient detail on metric validation, inter-rater reliability for graders, or systematic error analysis of task difficulty and multi-modal handling; this leaves the soundness of the performance claims dependent on unexamined setup choices.
minor comments (2)
- Ensure consistent model naming and versioning (e.g., 'Gemini-3.1-Pro' and 'GPT-5.4') across tables, figures, and text.
- Figure captions and table headers should explicitly state the normalization range and any per-domain adjustments to aid reader interpretation of the 0-1 scores.
Simulated Author's Rebuttal
We are grateful to the referee for their insightful comments, which have helped us identify areas for improvement in the BioXArena manuscript. We address the two major comments point by point below, committing to revisions that enhance the description of our methods without altering the core findings.
read point-by-point responses
-
Referee: [Abstract and §4] Abstract and §4 (Metrics and Evaluation): The headline average scores (0.666 and 0.636) and the claim that 'no single agent consistently dominates across all domains' rest on cross-domain comparability of biology-aware metrics normalized to [0,1]. The manuscript provides no explicit description of the normalization procedure, shared statistical grounding, expert baselines, or handling of differing difficulty floors across domains (e.g., AUC thresholds in imaging versus stricter biology-specific criteria in omics). If normalization is performed independently per task without calibration, domain scores become incomparable and the reported averages lose interpretability.
Authors: The referee raises a valid concern regarding the lack of explicit detail on metric normalization, which is crucial for interpreting the cross-domain average scores. We agree that this omission could undermine the comparability claims. In the revised manuscript, we will add a new subsection in §4 titled 'Normalization Procedure' that explicitly describes how each metric is scaled to [0,1]. Specifically, we normalize using task-specific lower and upper bounds derived from baseline performances (random models for lower bound and reference ML pipelines for upper bound). We will also discuss the handling of domain-specific difficulty by referencing expert-defined thresholds and provide examples across domains. This will allow readers to better assess the validity of the averages and the 'no single agent dominates' claim. We believe this addition will resolve the issue. revision: yes
-
Referee: [§3] §3 (Task Curation and Validation): The curation of 76 tasks from primary sources into a unified framework with hidden labels and held-out graders is load-bearing for the central claim that the benchmark 'accurately and fairly measure[s] real-world agent performance.' The manuscript states the design but supplies insufficient detail on metric validation, inter-rater reliability for graders, or systematic error analysis of task difficulty and multi-modal handling; this leaves the soundness of the performance claims dependent on unexamined setup choices.
Authors: We appreciate the referee pointing out the need for more transparency in task curation and validation. While the original manuscript outlines the overall design, we concur that more specifics on validation would strengthen the paper. In the revision, we will expand §3 with additional details on how metrics were validated, including pilot testing with domain experts for biology-aware criteria. For graders, we will report the results of our internal consistency checks (noting that they are code-driven with limited human review). We will also add a systematic error analysis section, including difficulty stratification by modality and domain, based on preliminary agent failure rates. These changes will provide better support for the benchmark's fairness and accuracy claims. revision: yes
Circularity Check
No circularity: benchmark results are direct empirical evaluations on curated tasks
full rationale
The paper presents BioXArena as a new benchmark with 76 tasks curated from primary sources, using hidden labels and biology-aware metrics normalized to [0,1]. It reports direct performance scores from running 11 agent configurations on private test samples in a standardized environment. No equations, derivations, or predictions are claimed that reduce by construction to fitted parameters, self-definitions, or self-citation chains. The evaluation relies on external agent code execution and held-out graders, remaining self-contained against the provided tasks without any load-bearing reduction to inputs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Biomedical ML tasks can be standardized into executable code pipelines evaluated with hidden test sets and biology-aware metrics on a 0-1 scale.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquationwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
BioXArena contains 76 end-to-end tasks across 9 domains... biology-aware metrics normalized to a 0 to 1 scale
-
IndisputableMonolith/Foundation/RealityFromDistinctionreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Agents are required to write executable code, train predictive models, and generate submissions
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
ReAct: Synergizing Reasoning and Acting in Language Models
Shunyu Yao, Jeffrey Zhao, Dian Yu, Nan Du, Izhak Shafran, Karthik Narasimhan, and Yuan Cao. React: Synergizing reasoning and acting in language models.arXiv preprint arXiv:2210.03629, 2022
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[2]
AgentBench: Evaluating LLMs as Agents
Xiao Liu, Hao Yu, Hanchen Zhang, Yifan Xu, Xuanyu Lei, Hanyu Lai, Yu Gu, Hangliang Ding, Kaiwen Men, Kejuan Yang, et al. Agentbench: Evaluating llms as agents.arXiv preprint arXiv:2308.03688, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[3]
Executable code actions elicit better llm agents
Xingyao Wang, Yangyi Chen, Lifan Yuan, Yizhe Zhang, Yunzhu Li, Hao Peng, and Heng Ji. Executable code actions elicit better llm agents. InForty-first International Conference on Machine Learning, 2024
work page 2024
-
[4]
Large language model-based data science agent: A survey.arXiv preprint arXiv:2508.02744, 2025
Ke Chen, Peiran Wang, Yaoning Yu, Xianyang Zhan, and Haohan Wang. Large language model-based data science agent: A survey.arXiv preprint arXiv:2508.02744, 2025
-
[5]
Biomni: A general-purpose biomedical ai agent.biorxiv, 2025
Kexin Huang, Serena Zhang, Hanchen Wang, Yuanhao Qu, Yingzhou Lu, Yusuf Roohani, Ryan Li, Lin Qiu, Gavin Li, Junze Zhang, et al. Biomni: A general-purpose biomedical ai agent.biorxiv, 2025
work page 2025
-
[6]
Stella: Self-evolving llm agent for biomedical research.arXiv preprint arXiv:2507.02004, 2025
Ruofan Jin, Zaixi Zhang, Mengdi Wang, and Le Cong. Stella: Self-evolving llm agent for biomedical research.arXiv preprint arXiv:2507.02004, 2025
-
[7]
LAB-Bench: Measuring Capabilities of Language Models for Biology Research
Jon M Laurent, Joseph D Janizek, Michael Ruzo, Michaela M Hinks, Michael J Hammerling, Siddharth Narayanan, Manvitha Ponnapati, Andrew D White, and Samuel G Rodriques. Lab-bench: Measuring capabilities of language models for biology research.arXiv preprint arXiv:2407.10362, 2024
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[8]
Ludovico Mitchener, Jon M Laurent, Alex Andonian, Benjamin Tenmann, Siddharth Narayanan, Geemi P Wellawatte, Andrew White, Lorenzo Sani, and Samuel G Rodriques. Bixbench: a comprehensive benchmark for llm-based agents in computational biology.arXiv preprint arXiv:2503.00096, 2025
-
[9]
BioAgent Bench: An AI Agent Evaluation Suite for Bioinformatics
Dionizije Fa, Marko ˇCuljak, Bruno Pandža, and Mateo ˇCupi´c. Bioagent bench: An ai agent evaluation suite for bioinformatics.arXiv preprint arXiv:2601.21800, 2026
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[10]
Yuyang Liu, Liuzhenghao Lv, Xiancheng Zhang, Jingya Wang Li Yuan, and Yonghong Tian. Bioprobench: Comprehensive dataset and benchmark in biological protocol understanding and reasoning.arXiv preprint arXiv:2505.07889, 2025
-
[11]
BiomniBench: Evaluating AI agents in biology
Phylo Team. BiomniBench: Evaluating AI agents in biology. Phylo Blog, 2026. URL https://phylo.bio/blog/evaluating-ai-agents-in-biology . Trace-based evaluation framework for biology agents; preliminary release: 15 data-analysis tasks (Biomni-DA-v0)
work page 2026
-
[12]
MLE-bench: Evaluating Machine Learning Agents on Machine Learning Engineering
Jun Shern Chan, Neil Chowdhury, Oliver Jaffe, James Aung, Dane Sherburn, Evan Mays, Giulio Starace, Kevin Liu, Leon Maksin, Tejal Patwardhan, et al. Mle-bench: Evaluating machine learning agents on machine learning engineering.arXiv preprint arXiv:2410.07095, 2024
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[13]
Alisia Lupidi, Bhavul Gauri, Thomas Simon Foster, Bassel Al Omari, Despoina Magka, Alberto Pepe, Alexis Audran-Reiss, Muna Aghamelu, Nicolas Baldwin, Lucia Cipolina-Kun, et al. Airs-bench: a suite of tasks for frontier ai research science agents.arXiv preprint arXiv:2602.06855, 2026
-
[14]
Bioml-bench: Evalua- tion of ai agents for end-to-end biomedical ml.bioRxiv, pages 2025–09, 2025
Henry E Miller, Matthew Greenig, Benjamin Tenmann, and Bo Wang. Bioml-bench: Evalua- tion of ai agents for end-to-end biomedical ml.bioRxiv, pages 2025–09, 2025. 10
work page 2025
-
[15]
GPT-5.4.https://openai.com/index/introducing-gpt-5-4/, 2026
OpenAI. GPT-5.4.https://openai.com/index/introducing-gpt-5-4/, 2026
work page 2026
-
[16]
Anthropic. Claude Opus 4.6. https://www.anthropic.com/news/claude-opus-4-6 , 2026
work page 2026
-
[17]
Alibaba Unveils Qwen3.6-Plus to Accelerate Agen- tic AI Deployment
Alibaba Cloud. Alibaba Unveils Qwen3.6-Plus to Accelerate Agen- tic AI Deployment. https://www.alibabacloud.com/press-room/ alibaba-unveils-qwen3-6-plus-to-accelerate-agentic, 2026
work page 2026
-
[18]
Gemini 3.1 Pro: A smarter model for your most complex tasks
Google. Gemini 3.1 Pro: A smarter model for your most complex tasks. https: //blog.google/innovation-and-ai/models-and-research/gemini-models/ gemini-3-1-pro/, 2026
work page 2026
-
[19]
GLM-5.1.https://docs.z.ai/guides/llm/glm-5.1, 2026
Z.AI. GLM-5.1.https://docs.z.ai/guides/llm/glm-5.1, 2026
work page 2026
-
[20]
Google DeepMind. Gemma 4 31B model. https://huggingface.co/google/ gemma-4-31B-it, 2026
work page 2026
-
[21]
DeepSeek-V3.2: Pushing the Frontier of Open Large Language Models
Aixin Liu, Aoxue Mei, Bangcai Lin, Bing Xue, Bingxuan Wang, Bingzheng Xu, Bochao Wu, Bowei Zhang, Chaofan Lin, Chen Dong, et al. Deepseek-v3. 2: Pushing the frontier of open large language models.arXiv preprint arXiv:2512.02556, 2025
work page internal anchor Pith review Pith/arXiv arXiv 2025
- [22]
-
[23]
Xinyu Zhu, Yuzhu Cai, Zexi Liu, Bingyang Zheng, Cheng Wang, Rui Ye, Jiaao Chen, Hanrui Wang, Wei-Chen Wang, Yuzhi Zhang, et al. Toward ultra-long-horizon agentic science: Cognitive accumulation for machine learning engineering.arXiv preprint arXiv:2601.10402, 2026
-
[24]
Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ron- neberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
work page 2021
-
[25]
Minkyung Baek, Frank DiMaio, Ivan Anishchenko, Justas Dauparas, Sergey Ovchinnikov, Gyu Rie Lee, Jue Wang, Qian Cong, Lisa N Kinch, R Dustin Schaeffer, et al. Accurate prediction of protein structures and interactions using a three-track neural network.Science, 373(6557):871–876, 2021
work page 2021
-
[26]
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proceedings of the national academy of sciences, 118(15):e2016239118, 2021
work page 2021
-
[27]
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
work page 2023
-
[28]
De novo design of protein structure and function with rfdiffusion.Nature, 620(7976):1089–1100, 2023
Joseph L Watson, David Juergens, Nathaniel R Bennett, Brian L Trippe, Jason Yim, Helen E Eisenach, Woody Ahern, Andrew J Borst, Robert J Ragotte, Lukas F Milles, et al. De novo design of protein structure and function with rfdiffusion.Nature, 620(7976):1089–1100, 2023
work page 2023
-
[29]
Jun Cheng, Guido Novati, Joshua Pan, Clare Bycroft, Akvil˙e Žemgulyt ˙e, Taylor Applebaum, Alexander Pritzel, Lai Hong Wong, Michal Zielinski, Tobias Sargeant, et al. Accurate proteome- wide missense variant effect prediction with alphamissense.Science, 381(6664):eadg7492, 2023
work page 2023
-
[30]
Effective gene expression prediction from sequence by integrating long-range interactions
Žiga Avsec, Vikram Agarwal, Daniel Visentin, Joseph R Ledsam, Agnieszka Grabska- Barwinska, Kyle R Taylor, Yannis Assael, John Jumper, Pushmeet Kohli, and David R Kelley. Effective gene expression prediction from sequence by integrating long-range interactions. Nature methods, 18(10):1196–1203, 2021. 11
work page 2021
-
[31]
Transfer learning enables predictions in network biology.Nature, 618(7965):616–624, 2023
Christina V Theodoris, Ling Xiao, Anant Chopra, Mark D Chaffin, Zeina R Al Sayed, Matthew C Hill, Helene Mantineo, Elizabeth M Brydon, Zexian Zeng, X Shirley Liu, et al. Transfer learning enables predictions in network biology.Nature, 618(7965):616–624, 2023
work page 2023
-
[32]
scgpt: toward building a foundation model for single-cell multi-omics using generative ai
Haotian Cui, Chloe Wang, Hassaan Maan, Kuan Pang, Fengning Luo, Nan Duan, and Bo Wang. scgpt: toward building a foundation model for single-cell multi-omics using generative ai. Nature methods, 21(8):1470–1480, 2024
work page 2024
-
[33]
Moritz Schaefer, Peter Peneder, Daniel Malzl, Salvo Danilo Lombardo, Mihaela Peycheva, Jake Burton, Anna Hakobyan, Varun Sharma, Thomas Krausgruber, Celine Sin, et al. Multimodal learning enables chat-based exploration of single-cell data.Nature Biotechnology, pages 1–11, 2025
work page 2025
-
[34]
Jinhyuk Lee, Wonjin Yoon, Sungdong Kim, Donghyeon Kim, Sunkyu Kim, Chan Ho So, and Jaewoo Kang. Biobert: a pre-trained biomedical language representation model for biomedical text mining.Bioinformatics, 36(4):1234–1240, 2020
work page 2020
-
[35]
Yu Gu, Robert Tinn, Hao Cheng, Michael Lucas, Naoto Usuyama, Xiaodong Liu, Tristan Naumann, Jianfeng Gao, and Hoifung Poon. Domain-specific language model pretraining for biomedical natural language processing.ACM Transactions on Computing for Healthcare (HEALTH), 3(1):1–23, 2021
work page 2021
-
[36]
Richard J Chen, Tong Ding, Ming Y Lu, Drew FK Williamson, Guillaume Jaume, Andrew H Song, Bowen Chen, Andrew Zhang, Daniel Shao, Muhammad Shaban, et al. Towards a general-purpose foundation model for computational pathology.Nature medicine, 30(3): 850–862, 2024
work page 2024
-
[37]
Moleculenet: a benchmark for molecular machine learning.Chemical science, 9(2):513–530, 2018
Zhenqin Wu, Bharath Ramsundar, Evan N Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S Pappu, Karl Leswing, and Vijay Pande. Moleculenet: a benchmark for molecular machine learning.Chemical science, 9(2):513–530, 2018
work page 2018
-
[38]
Kexin Huang, Tianfan Fu, Wenhao Gao, Yue Zhao, Yusuf Roohani, Jure Leskovec, Con- nor W Coley, Cao Xiao, Jimeng Sun, and Marinka Zitnik. Therapeutics data commons: Machine learning datasets and tasks for drug discovery and development.arXiv preprint arXiv:2102.09548, 2021
-
[39]
Malte D Luecken, Scott Gigante, Daniel B Burkhardt, Robrecht Cannoodt, Daniel C Strobl, Nikolay S Markov, Luke Zappia, Giovanni Palla, Wesley Lewis, Daniel Dimitrov, et al. Defining and benchmarking open problems in single-cell analysis.Nature Biotechnology, 43 (7):1035–1040, 2025
work page 2025
-
[40]
Christopher Lance, Malte D Luecken, Daniel B Burkhardt, Robrecht Cannoodt, Pia Rauten- strauch, Anna Laddach, Aidyn Ubingazhibov, Zhi-Jie Cao, Kaiwen Deng, Sumeer Khan, et al. Multimodal single cell data integration challenge: results and lessons learned.BioRxiv, pages 2022–04, 2022
work page 2022
-
[41]
Pascal Notin, Aaron Kollasch, Daniel Ritter, Lood Van Niekerk, Steffanie Paul, Han Spinner, Nathan Rollins, Ada Shaw, Rose Orenbuch, Ruben Weitzman, et al. Proteingym: Large- scale benchmarks for protein fitness prediction and design.Advances in neural information processing systems, 36:64331–64379, 2023
work page 2023
-
[42]
Polaris: The benchmark platform for drug discovery
Polaris consortium. Polaris: The benchmark platform for drug discovery. https:// polarishub.io, 2024
work page 2024
-
[43]
Xingbo Du, Loka Li, Duzhen Zhang, and Le Song. Memr3: Memory retrieval via reflective reasoning for llm agents.arXiv preprint arXiv:2512.20237, 2025
-
[44]
Aide: Human-level performance in data science competitions, 2024
Dominik Schmidt, Yuxiang Wu, and Zhengyao Jiang. Aide: Human-level performance in data science competitions, 2024
work page 2024
-
[45]
Qian Huang, Jian V ora, Percy Liang, and Jure Leskovec. Mlagentbench: Evaluating language agents on machine learning experimentation.arXiv preprint arXiv:2310.03302, 2023. 12
-
[46]
A Survey on Code Generation with LLM-based Agents
Yihong Dong, Xue Jiang, Jiaru Qian, Tian Wang, Kechi Zhang, Zhi Jin, and Ge Li. A survey on code generation with llm-based agents.arXiv preprint arXiv:2508.00083, 2025
work page internal anchor Pith review arXiv 2025
-
[47]
GTEx Consortium. The gtex consortium atlas of genetic regulatory effects across human tissues.Science, 369(6509):1318–1330, 2020
work page 2020
-
[48]
An integrated encyclopedia of dna elements in the human genome.Nature, 489(7414):57, 2012
ENCODE Project Consortium et al. An integrated encyclopedia of dna elements in the human genome.Nature, 489(7414):57, 2012
work page 2012
-
[49]
A reference map of the human binary protein interactome.Nature, 580(7803):402–408, 2020
Katja Luck, Dae-Kyum Kim, Luke Lambourne, Kerstin Spirohn, Bridget E Begg, Wenting Bian, Ruth Brignall, Tiziana Cafarelli, Francisco J Campos-Laborie, Benoit Charloteaux, et al. A reference map of the human binary protein interactome.Nature, 580(7803):402–408, 2020
work page 2020
-
[50]
Cath–a hierarchic classification of protein domain structures.Structure, 5 (8):1093–1109, 1997
Christine A Orengo, Alex D Michie, Susan Jones, David T Jones, Mark B Swindells, and Janet M Thornton. Cath–a hierarchic classification of protein domain structures.Structure, 5 (8):1093–1109, 1997
work page 1997
-
[51]
Nicole Lambert, Alex Robertson, Mohini Jangi, Sean McGeary, Phillip A Sharp, and Christo- pher B Burge. Rna bind-n-seq: quantitative assessment of the sequence and structural binding specificity of rna binding proteins.Molecular cell, 54(5):887–900, 2014
work page 2014
-
[52]
Structural imprints in vivo decode rna regulatory mechanisms.Nature, 519(7544):486–490, 2015
Robert C Spitale, Ryan A Flynn, Qiangfeng Cliff Zhang, Pete Crisalli, Byron Lee, Jong-Wha Jung, Hannes Y Kuchelmeister, Pedro J Batista, Eduardo A Torre, Eric T Kool, et al. Structural imprints in vivo decode rna regulatory mechanisms.Nature, 519(7544):486–490, 2015
work page 2015
-
[53]
Clinvar: improvements to accessing data.Nucleic acids research, 48(D1):D835–D844, 2020
Melissa J Landrum, Shanmuga Chitipiralla, Garth R Brown, Chao Chen, Baoshan Gu, Jennifer Hart, Douglas Hoffman, Wonhee Jang, Kuljeet Kaur, Chunlei Liu, et al. Clinvar: improvements to accessing data.Nucleic acids research, 48(D1):D835–D844, 2020
work page 2020
-
[54]
Andrew JC Russell, Jackson A Weir, Naeem M Nadaf, Matthew Shabet, Vipin Kumar, Sandeep Kambhampati, Ruth Raichur, Giovanni J Marrero, Sophia Liu, Karol S Balderrama, et al. Slide-tags enables single-nucleus barcoding for multimodal spatial genomics.Nature, 625 (7993):101–109, 2024
work page 2024
-
[55]
Marlon Stoeckius, Christoph Hafemeister, William Stephenson, Brian Houck-Loomis, Pratip K Chattopadhyay, Harold Swerdlow, Rahul Satija, and Peter Smibert. Simultaneous epitope and transcriptome measurement in single cells.Nature methods, 14(9):865–868, 2017
work page 2017
-
[56]
Single cell dual-omic atlas of the human developing retina.Nature Communications, 15(1):6792, 2024
Zhen Zuo, Xuesen Cheng, Salma Ferdous, Jianming Shao, Jin Li, Yourong Bao, Jean Li, Jiaxiong Lu, Antonio Jacobo Lopez, Juliette Wohlschlegel, et al. Single cell dual-omic atlas of the human developing retina.Nature Communications, 15(1):6792, 2024
work page 2024
-
[57]
Andriy Kryshtafovych, Torsten Schwede, Maya Topf, Krzysztof Fidelis, and John Moult. Critical assessment of methods of protein structure prediction (casp)—round xiv.Proteins: Structure, Function, and Bioinformatics, 89(12):1607–1617, 2021
work page 2021
-
[58]
The protein data bank.Nucleic acids research, 28(1):235–242, 2000
Helen M Berman, John Westbrook, Zukang Feng, Gary Gilliland, Talapady N Bhat, Helge Weissig, Ilya N Shindyalov, and Philip E Bourne. The protein data bank.Nucleic acids research, 28(1):235–242, 2000
work page 2000
-
[59]
John-Marc Chandonia, Naomi K Fox, and Steven E Brenner. Scope: manual curation and artifact removal in the structural classification of proteins–extended database.Journal of molecular biology, 429(3):348–355, 2017
work page 2017
-
[60]
Pdb-wide collection of binding data: current status of the pdbbind database
Zhihai Liu, Yan Li, Li Han, Jie Li, Jie Liu, Zhixiong Zhao, Wei Nie, Yuchen Liu, and Renxiao Wang. Pdb-wide collection of binding data: current status of the pdbbind database. Bioinformatics, 31(3):405–412, 2015
work page 2015
-
[61]
Janet Piñero, Juan Manuel Ramírez-Anguita, Josep Saüch-Pitarch, Francesco Ronzano, Emilio Centeno, Ferran Sanz, and Laura I Furlong. The disgenet knowledge platform for disease genomics: 2019 update.Nucleic acids research, 48(D1):D845–D855, 2020
work page 2019
-
[62]
Gene ontology: tool for the unification of biology.Nature genetics, 25(1):25–29, 2000
Michael Ashburner, Catherine A Ball, Judith A Blake, David Botstein, Heather Butler, J Michael Cherry, Allan P Davis, Kara Dolinski, Selina S Dwight, Janan T Eppig, et al. Gene ontology: tool for the unification of biology.Nature genetics, 25(1):25–29, 2000. 13
work page 2000
-
[63]
Minoru Kanehisa, Miho Furumichi, Yoko Sato, Masayuki Kawashima, and Mari Ishiguro- Watanabe. Kegg for taxonomy-based analysis of pathways and genomes.Nucleic acids research, 51(D1):D587–D592, 2023
work page 2023
-
[64]
The reactome pathway knowledgebase.Nucleic acids research, 48(D1):D498–D503, 2020
Bijay Jassal, Lisa Matthews, Guilherme Viteri, Chuqiao Gong, Pascual Lorente, Antonio Fabregat, Konstantinos Sidiropoulos, Justin Cook, Marc Gillespie, Robin Haw, et al. The reactome pathway knowledgebase.Nucleic acids research, 48(D1):D498–D503, 2020
work page 2020
-
[65]
Damian Szklarczyk, Rebecca Kirsch, Mikaela Koutrouli, Katerina Nastou, Farrokh Mehryary, Radja Hachilif, Annika L Gable, Tao Fang, Nadezhda T Doncheva, Sampo Pyysalo, et al. The string database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest.Nucleic acids research, 51(D1):D638–D646, 2023
work page 2023
-
[66]
Madalina Giurgiu, Julian Reinhard, Barbara Brauner, Irmtraud Dunger-Kaltenbach, Gisela Fobo, Goar Frishman, Corinna Montrone, and Andreas Ruepp. Corum: the comprehensive resource of mammalian protein complexes—2019.Nucleic acids research, 47(D1):D559– D563, 2019
work page 2019
-
[67]
Jing Guo, Hui Liu, and Jie Zheng. Synlethdb: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets.Nucleic acids research, 44(D1):D1011–D1017, 2016
work page 2016
-
[68]
Michael K Gilson, Tiqing Liu, Michael Baitaluk, George Nicola, Linda Hwang, and Jenny Chong. Bindingdb in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology.Nucleic acids research, 44(D1):D1045–D1053, 2016
work page 2015
-
[69]
Srinivas Niranj Chandrasekaran, Jeanelle Ackerman, Eric Alix, D Michael Ando, John Arevalo, Melissa Bennion, Nicolas Boisseau, Adriana Borowa, Justin D Boyd, Laurent Brino, et al. Jump cell painting dataset: morphological impact of 136,000 chemical and genetic perturba- tions.BioRxiv, pages 2023–03, 2023
work page 2023
-
[70]
Sunghwan Kim, Jie Chen, Tiejun Cheng, Asta Gindulyte, Jia He, Siqian He, Qingliang Li, Benjamin A Shoemaker, Paul A Thiessen, Bo Yu, et al. Pubchem in 2021: new data content and improved web interfaces.Nucleic acids research, 49(D1):D1388–D1395, 2021
work page 2021
-
[71]
Chembl: towards direct deposition of bioassay data.Nucleic acids research, 47(D1):D930–D940, 2019
David Mendez, Anna Gaulton, A Patrícia Bento, Jon Chambers, Marleen De Veij, Eloy Félix, María Paula Magariños, Juan F Mosquera, Prudence Mutowo, Michał Nowotka, et al. Chembl: towards direct deposition of bioassay data.Nucleic acids research, 47(D1):D930–D940, 2019
work page 2019
-
[72]
Jane F Armstrong, Elena Faccenda, Simon D Harding, Adam J Pawson, Christopher Southan, Joanna L Sharman, Brice Campo, David R Cavanagh, Stephen PH Alexander, Anthony P Davenport, et al. The iuphar/bps guide to pharmacology in 2020: extending immunopharma- cology content and introducing the iuphar/mmv guide to malaria pharmacology.Nucleic acids research, 4...
work page 2020
-
[73]
Ruili Huang, Menghang Xia, Srilatha Sakamuru, Jinghua Zhao, Sampada A Shahane, Matias Attene-Ramos, Tongan Zhao, Christopher P Austin, and Anton Simeonov. Modelling the tox21 10 k chemical profiles for in vivo toxicity prediction and mechanism characterization.Nature communications, 7(1):10425, 2016
work page 2016
-
[74]
A landscape of pharmacogenomic interactions in cancer.Cell, 166(3):740–754, 2016
Francesco Iorio, Theo A Knijnenburg, Daniel J Vis, Graham R Bignell, Michael P Menden, Michael Schubert, Nanne Aben, Emanuel Gonçalves, Syd Barthorpe, Howard Lightfoot, et al. A landscape of pharmacogenomic interactions in cancer.Cell, 166(3):740–754, 2016
work page 2016
-
[75]
Joseph M Replogle, Reuben A Saunders, Angela N Pogson, Jeffrey A Hussmann, Alexander Lenail, Alina Guna, Lauren Mascibroda, Eric J Wagner, Karen Adelman, Gila Lithwick- Yanai, et al. Mapping information-rich genotype-phenotype landscapes with genome-scale perturb-seq.Cell, 185(14):2559–2575, 2022
work page 2022
-
[76]
Sanjay R Srivatsan, José L McFaline-Figueroa, Vijay Ramani, Lauren Saunders, Junyue Cao, Jonathan Packer, Hannah A Pliner, Dana L Jackson, Riza M Daza, Lena Christiansen, et al. Massively multiplex chemical transcriptomics at single-cell resolution.Science, 367(6473): 45–51, 2020. 14
work page 2020
-
[77]
Eleni P Mimitou, Anthony Cheng, Antonino Montalbano, Stephanie Hao, Marlon Stoeckius, Mateusz Legut, Timothy Roush, Alberto Herrera, Efthymia Papalexi, Zhengqing Ouyang, et al. Multiplexed detection of proteins, transcriptomes, clonotypes and crispr perturbations in single cells.Nature methods, 16(5):409–412, 2019
work page 2019
-
[78]
Aditya Pratapa, Amogh P Jalihal, Jeffrey N Law, Aditya Bharadwaj, and andT M Murali. Benchmarking algorithms for gene regulatory network inference from single-cell transcrip- tomic data.Nature methods, 17(2):147–154, 2020
work page 2020
-
[79]
Aravind Subramanian, Rajiv Narayan, Steven M Corsello, David D Peck, Ted E Natoli, Xiaodong Lu, Joshua Gould, John F Davis, Andrew A Tubelli, Jacob K Asiedu, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles.Cell, 171(6): 1437–1452, 2017
work page 2017
-
[80]
V olker Bergen, Marius Lange, Stefan Peidli, F Alexander Wolf, and Fabian J Theis. Generaliz- ing rna velocity to transient cell states through dynamical modeling.Nature biotechnology, 38 (12):1408–1414, 2020
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.