Strategies for Molecular Dynamics using Hybrid Systems: LAMMPS Use Case
Pith reviewed 2026-06-28 12:37 UTC · model grok-4.3
The pith
Hybrid MPI+OpenMP configurations in LAMMPS scale better than pure MPI for large-scale biomolecular simulations by reducing communication overhead.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
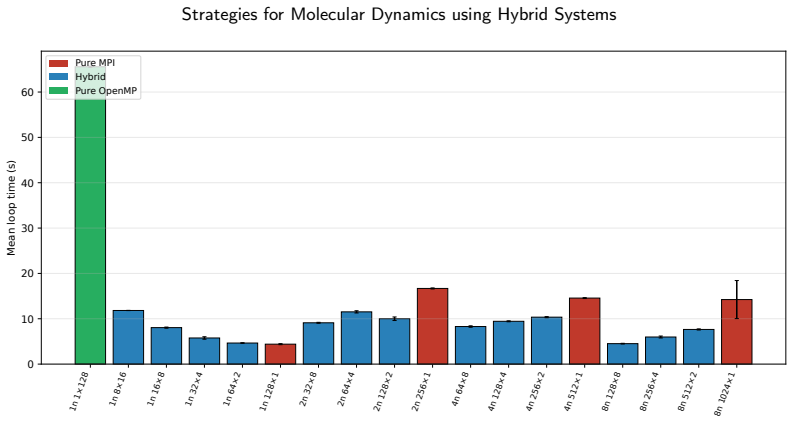
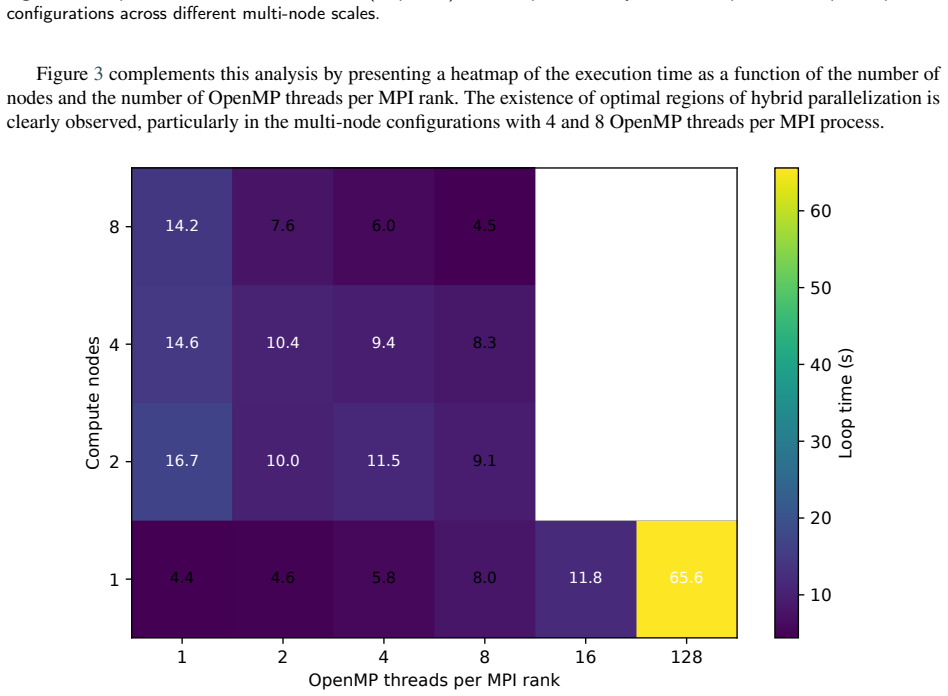
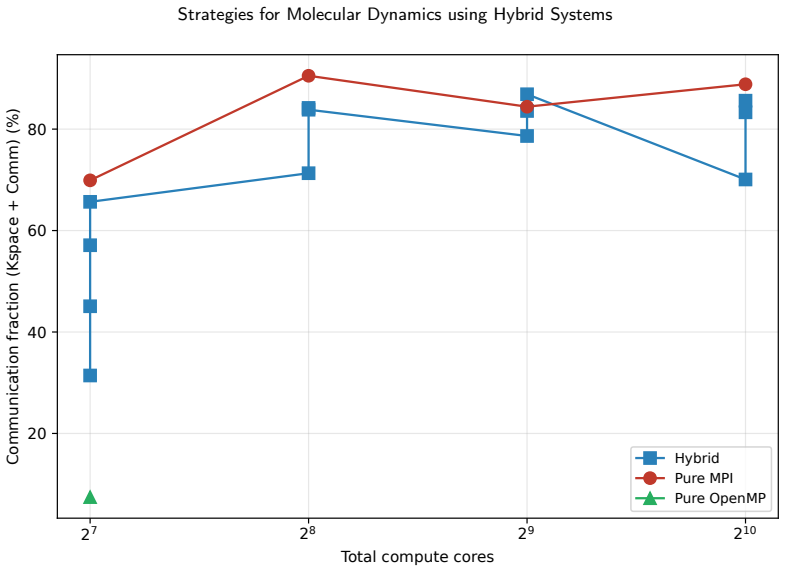
For the LAMMPS simulator applied to coarse-grained biomolecular dynamics of the Tritrpticin peptide, hybrid MPI+OpenMP parallelization delivers superior scalability on multi-node HPC systems compared to pure MPI because it lowers inter-process communication volume and better matches the NUMA memory hierarchy, leading to lower execution times at scales up to 1024 cores.
What carries the argument
The hybrid MPI+OpenMP execution model within LAMMPS, which combines message passing across nodes with shared-memory threading within nodes for the spatial domain decomposition.
If this is right
- Pure MPI executions show scalability degradation beyond single-node due to communication overhead.
- Hybrid configurations reduce the fraction of time spent in communication and electrostatic routines at large scales.
- Performance in these simulations depends on balancing parallelization granularity, spatial decomposition, and distributed communication costs.
- Hybrid MPI+OpenMP offers a more sustainable path for running coarse-grained biomolecular simulations on many-core HPC architectures.
Where Pith is reading between the lines
- Similar hybrid strategies could be tested in other molecular dynamics packages to see if the scalability benefit generalizes.
- The results on one peptide suggest checking whether the advantage holds for larger or more complex biomolecular systems.
- Future work might explore optimal thread-to-process ratios for different hardware NUMA configurations.
Load-bearing premise
The measured performance edge of hybrid over pure MPI on the Tritrpticin workload and the specific testbed hardware will apply to other biomolecular systems and different HPC machines.
What would settle it
A follow-up run of the same LAMMPS tests on a different peptide or protein system using the same hybrid versus pure MPI comparison at 1024 cores, checking whether the hybrid still shows lower execution time.
Figures






read the original abstract
The complexity of biomolecular simulations has substantially increased the demand for High-Performance Computing (HPC) infrastructures, particularly in molecular dynamics and coarse-grained modeling. This work presents a systematic performance and scalability analysis of the LAMMPS simulator for coarse-grained biomolecular simulations, using the antimicrobial peptide Tritrpticin (PDB ID: 1D6X) as the experimental workload. Pure MPI and hybrid MPI+OpenMP executions were evaluated in HPC environments comprising up to 8 compute nodes and 1024 simultaneous cores. Metrics of execution time, speedup, parallel efficiency, statistical variability, and internal time decomposition were investigated. Results showed that pure MPI executions deliver excellent performance in single-node environments but suffer scalability degradation in multi-node executions due to communication overhead and inter-process synchronization. Hybrid MPI+OpenMP configurations proved more efficient at large scale, reducing communication costs and better exploiting the NUMA memory hierarchy. The computational breakdown revealed that communication and electrostatic interaction routines accounted for the largest fraction of execution time at the largest pure-MPI scales. These results reinforce that performance of biomolecular HPC applications depends directly on the balance among parallelization granularity, spatial decomposition, and distributed communication costs. Hybrid MPI+OpenMP strategies represent a more sustainable alternative for coarse-grained biomolecular simulations on modern many-core architectures.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports a performance and scalability study of LAMMPS for coarse-grained biomolecular MD simulations using the Tritrpticin peptide (PDB 1D6X) as the sole workload. Pure MPI and hybrid MPI+OpenMP runs are compared on an HPC testbed with up to 8 nodes and 1024 cores, using metrics of execution time, speedup, parallel efficiency, statistical variability, and internal time decomposition. The central claim is that pure MPI scales well within a node but degrades at multi-node scales due to communication overhead, while hybrid MPI+OpenMP is superior at large scale by lowering communication costs and better exploiting the NUMA hierarchy, making hybrid strategies a more sustainable choice for such simulations.
Significance. If the trends hold beyond the reported case, the work supplies concrete empirical guidance on parallelization granularity for biomolecular HPC codes, with the internal time breakdown (highlighting communication and electrostatics as dominant at scale) being a useful diagnostic contribution. The systematic comparison of pure vs. hybrid on a real application is a strength for practitioners, though the single-workload design constrains broader applicability to other coarse-grained systems or hardware.
major comments (2)
- [Abstract] Abstract: the headline claim that hybrid MPI+OpenMP 'proved more efficient at large scale' and represents 'a more sustainable alternative for coarse-grained biomolecular simulations' is derived exclusively from the 1D6X peptide; no results are shown for other system sizes, force fields, or interaction profiles that would be needed to establish that the observed crossover in communication-to-compute ratio is representative rather than workload-specific.
- [Abstract] Abstract: although 'statistical variability' is listed among the investigated metrics, the text supplies no error bars, standard deviations, or hypothesis tests on the timing data, leaving the reported trends in execution time and efficiency without quantified uncertainty and making it impossible to judge whether observed differences exceed measurement noise.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We address each major comment below and indicate the revisions we will make.
read point-by-point responses
-
Referee: [Abstract] Abstract: the headline claim that hybrid MPI+OpenMP 'proved more efficient at large scale' and represents 'a more sustainable alternative for coarse-grained biomolecular simulations' is derived exclusively from the 1D6X peptide; no results are shown for other system sizes, force fields, or interaction profiles that would be needed to establish that the observed crossover in communication-to-compute ratio is representative rather than workload-specific.
Authors: We agree that the empirical results are derived from a single workload (the 1D6X Tritrpticin peptide). This system was chosen as a representative coarse-grained biomolecular case with typical interaction profiles, allowing detailed internal time decomposition. The observed communication overhead trends are expected to generalize to similar systems, but we accept that broader validation across system sizes and force fields would strengthen the claim. In revision we will qualify the abstract to state that hybrid MPI+OpenMP proved more efficient at large scale for this workload and add a note in the conclusions on the value of future multi-workload studies. revision: yes
-
Referee: [Abstract] Abstract: although 'statistical variability' is listed among the investigated metrics, the text supplies no error bars, standard deviations, or hypothesis tests on the timing data, leaving the reported trends in execution time and efficiency without quantified uncertainty and making it impossible to judge whether observed differences exceed measurement noise.
Authors: The manuscript does list statistical variability as a metric and performed repeated runs to assess it, yet the reported figures and text omit explicit error bars or standard deviations. This is a reporting omission that prevents readers from evaluating whether differences exceed noise. We will revise the manuscript to include error bars (standard deviation across runs) on all timing and efficiency plots and add a brief discussion of the observed variability. revision: yes
Circularity Check
No circularity: purely empirical timing measurements on one workload with no derivations or self-referential equations
full rationale
The paper reports benchmark timings, speedups, efficiency, and time breakdowns for LAMMPS runs of the 1D6X peptide under pure-MPI vs. hybrid MPI+OpenMP on a fixed testbed. No equations, fitted parameters, predictions derived from inputs, or self-citation chains appear in the abstract or described methodology. All claims rest on direct measurement rather than any reduction to prior results or definitions within the paper. This matches the reader's assessment of score 0.0 and contains none of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The Tritrpticin coarse-grained simulation and the tested HPC nodes are representative of typical biomolecular workloads and modern many-core systems.
Forward citations
Cited by 1 Pith paper
-
How far does a random forest generalize from a 54-run LAMMPS+SPICA benchmark?
A random forest surrogate trained on 54 LAMMPS runs achieves 4% relative error on loop time and ranks configurations correctly within hardware regimes but degrades across architectural boundaries.
Reference graph
Works this paper leans on
-
[1]
T.Liu,D.Lu,H.Zhang,M.Zheng,H.Yang,Y.Xu,C.Luo,W.Zhu,K.Yu,H.Jiang,Applyinghigh-performancecomputingindrugdiscovery and molecular simulation, National Science Review 3 (1) (2016) 49–63.doi:10.1093/nsr/nww003
-
[2]
S. A. Hollingsworth, R. O. Dror, Molecular Dynamics Simulation for All, Neuron 99 (6) (2018) 1129–1143.doi:10.1016/j.neuron. 2018.08.011
-
[3]
B. Acun, D. J. Hardy, L. V. Kale, K. Li, J. C. Phillips, J. E. Stone, Scalable molecular dynamics with NAMD on the Summit system, IBM Journal of Research and Development 62 (6) (2018) 4:1–4:9.doi:10.1147/jrd.2018.2888986
-
[4]
C. Kutzner, V. Miletić, K. Palacio Rodríguez, M. Rampp, G. Hummer, B. L. de Groot, H. Grubmüller, Scaling of the GROMACS Molecular Dynamics Code to 65k CPU Cores on an HPC Cluster, Journal of Computational Chemistry 46 (5) (Feb. 2025).doi:10.1002/jcc.70059
-
[5]
M.Koch,C.Arlandini,G.Antonopoulos,A.Baretta,P.Beaujean,G.J.Bex,M.E.Biancolini,S.Celi,E.Costa,L.Drescher,V.Eleftheriadis, N.A.Fadel,A.Fink,F.Galbiati,I.Hatzakis,G.Hompis,N.Lewandowski,A.Memmolo,C.Mensch,D.Obrist,V.Paneta,P.Papadimitroulas, K. Petropoulos, S. Porziani, G. Savvidis, K. Sethia, P. Strakos, P. Svobodova, E. Vignali, HPC+ in the medical field: ...
-
[6]
Z. D. Stephens, S. Y. Lee, F. Faghri, R. H. Campbell, C. Zhai, M. J. Efron, R. Iyer, M. C. Schatz, S. Sinha, G. E. Robinson, Big Data: Astronomical or Genomical?, PLOS Biology 13 (7) (2015) e1002195.doi:10.1371/journal.pbio.1002195
-
[7]
G.Lightbody,V.Haberland,F.Browne,L.Taggart,H.Zheng,E.Parkes,J.K.Blayney,Reviewofapplicationsofhigh-throughputsequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application, Briefings in Bioinformatics 20 (5) (2019) 1795–1811.doi:10.1093/bib/bby051
-
[8]
J. Dean, S. Ghemawat, MapReduce: Simplified data processing on large clusters, Communications of the ACM 51 (1) (2008) 107–113. doi:10.1145/1327452.1327492
-
[9]
G. Goth, Eric Brewer: Change agent, Communications of the ACM 53 (7) (2010) 24–24.doi:10.1145/1785414.1785425
-
[10]
P. Kovatch, L. Gai, H. M. Cho, E. Fluder, D. Jiang, Optimizing High-Performance Computing Systems for Biomedical Workloads, in: 2020 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW), IEEE, 2020, pp. 183–192.doi:10.1109/ ipdpsw50202.2020.00040
arXiv 2020
-
[11]
J. Li, S. Wang, S. Rudinac, A. Osseyran, High-performance computing in healthcare: An automatic literature analysis perspective, Journal of Big Data 11 (1) (May 2024).doi:10.1186/s40537-024-00929-2
-
[12]
S.Y.Mashayak,M.N.Jochum,K.Koschke,N.R.Aluru,V.Rühle,C.Junghans,RelativeEntropyandOptimization-DrivenCoarse-Graining Methods in VOTCA, PLOS ONE 10 (7) (2015) e0131754.doi:10.1371/journal.pone.0131754
-
[13]
P. R. Alliata, D. Rubaga, D. Kumlin, A. Puliga, Parallelizing Drug Discovery: HPC Pipelines for Alzheimer’s Molecular Docking and Simulation (2025).doi:10.48550/ARXIV.2509.00937
-
[14]
S.J.Marrink,D.P.Tieleman,PerspectiveontheMartinimodel,ChemicalSocietyReviews42(16)(2013)6801.doi:10.1039/c3cs60093a
-
[15]
E. Braun, J. Gilmer, H. B. Mayes, D. L. Mobley, J. I. Monroe, S. Prasad, D. M. Zuckerman, Best Practices for Foundations in Molecular Simulations [Article v1.0], Living Journal of Computational Molecular Science 1 (1) (2018) 5957.doi:10.33011/livecoms.1.1.5957
-
[16]
E. R. Georgieva, Protein Conformational Dynamics upon Association with the Surfaces of Lipid Membranes and Engineered Nanoparticles: Insights from Electron Paramagnetic Resonance Spectroscopy, Molecules 25 (22) (2020) 5393.doi:10.3390/molecules25225393
-
[17]
D. Ramirez, V. Kohar, M. Lu, Toward Modeling Context-Specific EMT Regulatory Networks Using Temporal Single Cell RNA-Seq Data, Frontiers in Molecular Biosciences 7 (Apr. 2020).doi:10.3389/fmolb.2020.00054
-
[18]
J.Herber,J.Njavro,R.Feederle,U.Schepers,U.C.Müller,S.Bräse,S.A.Müller,S.F.Lichtenthaler,ClickChemistry-mediatedBiotinylation RevealsaFunctionfortheProteaseBACE1inModulatingtheNeuronalSurfaceGlycoproteome,Molecular&CellularProteomics17(8) (2018) 1487–1501.doi:10.1074/mcp.ra118.000608
-
[19]
G. Adrian, E. Konradsson, M. Lempart, S. Bäck, C. Ceberg, K. Petersson, The FLASH effect depends on oxygen concentration, The British Journal of Radiology 93 (1106) (Dec. 2019).doi:10.1259/bjr.20190702. P. H. L. Ramalho et al.:Preprint submitted to ElsevierPage 17 of 18 Strategies for Molecular Dynamics using Hybrid Systems
-
[20]
S. Seo, W. Shinoda, SPICA Force Field for Lipid Membranes: Domain Formation Induced by Cholesterol, Journal of Chemical Theory and Computation 15 (1) (2018) 762–774.doi:10.1021/acs.jctc.8b00987
-
[21]
L. Monticelli, D. P. Tieleman, Force Fields for Classical Molecular Dynamics, in: Biomolecular Simulations, Humana Press, 2012, pp. 197– 213.doi:10.1007/978-1-62703-017-5_8
-
[22]
R. O. Dror, R. M. Dirks, J. Grossman, H. Xu, D. E. Shaw, Biomolecular Simulation: A Computational Microscope for Molecular Biology, Annual Review of Biophysics 41 (1) (2012) 429–452.doi:10.1146/annurev-biophys-042910-155245
-
[23]
K. Lindorff-Larsen, P. Maragakis, S. Piana, M. P. Eastwood, R. O. Dror, D. E. Shaw, Systematic Validation of Protein Force Fields against Experimental Data, PLoS ONE 7 (2) (2012) e32131.doi:10.1371/journal.pone.0032131
-
[24]
J. Gelpi, A. Hospital, R. Goñi, M. Orozco, Molecular dynamics simulations: Advances and applications, Advances and Applications in Bioinformatics and Chemistry (2015) 37doi:10.2147/aabc.s70333
-
[25]
K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov, A. D. Mackerell, CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, Journal of Computational Chemistry 31 (4) (2009) 671–690.doi:10.1002/jcc.21367
-
[26]
Y.Nochez,S.Arsene,N.Gueguen,A.Chevrollier,M.Ferré,V.Guillet,etal.,Acuteandlate-onsetopticatrophyduetoanovelOPA1mutation leading to a mitochondrial coupling defect, Molecular Vision 15 (2009) 598–608
2009
-
[27]
M. C. Righetti, L. Aliotta, N. Mallegni, M. Gazzano, E. Passaglia, P. Cinelli, A. Lazzeri, Constrained Amorphous Interphase and Mechanical Properties of Poly(3-Hydroxybutyrate-co-3-Hydroxyvalerate), Frontiers in Chemistry 7 (Nov. 2019).doi:10.3389/fchem.2019.00790
-
[28]
Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J
S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, Journal of Computational Physics 117 (1) (1995) 1–19. doi:10.1006/jcph.1995.1039
-
[29]
U.Essmann,L.Perera,M.L.Berkowitz,T.Darden,H.Lee,L.G.Pedersen,AsmoothparticlemeshEwaldmethod,TheJournalofChemical Physics 103 (19) (1995) 8577–8593.doi:10.1063/1.470117
-
[30]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T.D.Nguyen,R.Shan,M.J.Stevens,J.Tranchida,C.Trott,S.J.Plimpton,LAMMPS-aflexiblesimulationtoolforparticle-basedmaterials modelingattheatomic,meso,andcontinuumscales,ComputerPhysicsCommunications271(2022)108171.doi:10.1016/j...
-
[31]
A. Rohskopf, C. Sievers, N. Lubbers, M. A. Cusentino, J. Goff, J. Janssen, M. McCarthy, D. M. de Oca Zapiain, S. Nikolov, K. Sargsyan, D. Sema, E. Sikorski, L. Williams, A. P. Thompson, M. A. Wood, FitSNAP: Atomistic machine learning with LAMMPS, Journal of Open Source Software 8 (84) (2023) 5118.doi:10.21105/joss.05118
-
[32]
G. M. Amdahl, Validity of the single processor approach to achieving large scale computing capabilities, in: Proceedings of the April 18-20, 1967,SpringJointComputerConferenceon-AFIPS’67(Spring),AFIPS’67(Spring),ACMPress,1967,p.483.doi:10.1145/1465482. 1465560
-
[33]
K. Ghedira, O. Khamessi, C. Hkimi, S. Kamoun, N. Dhamer, K. Daassi, W. Ben Salah, H. Othman, W. Belhadj, Y. Ghorbal, Design and implementation of a scalable high-performance computing (HPC) cluster for omics data analysis: Achievements, challenges and recommendations in LMICs, GigaScience 13 (2024).doi:10.1093/gigascience/giae060
-
[34]
Y. Miyazaki, Y. Teppei, W. Shinoda, onefive13, SPICA-group/spica-tools: V1.0.0, Zenodo (Feb. 2024).doi:10.5281/ZENODO.10611578. P. H. L. Ramalho et al.:Preprint submitted to ElsevierPage 18 of 18
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.