Structure-Aware Prediction of PROTAC-Mediated Protein Degradability via Graph Neural Networks
Pith reviewed 2026-06-28 11:57 UTC · model grok-4.3
The pith
DegradoMap predicts PROTAC degradability from protein structure and E3 ligase identity alone using a graph neural network.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
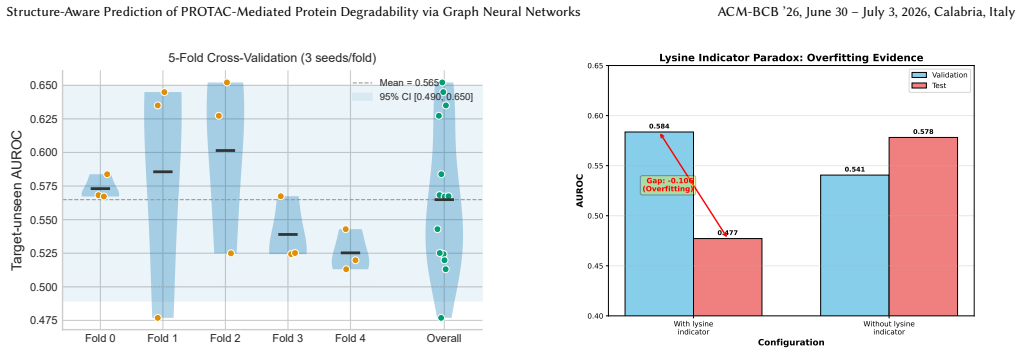
DegradoMap encodes protein structures via lysine-weighted graph pooling with per-protein normalization, models protein-E3 compatibility through cross-attention, and incorporates Cancer Dependency Map context to output a degradability probability. On the PROTAC-8K benchmark of 3,101 samples spanning 155 targets and 10 E3 ligases, the model records 0.646 plus or minus 0.124 AUROC under target-unseen splits and 0.811 AUROC under CRBN-to-VHL E3 transfer, surpassing GNN and ML baselines, while achieving 74 percent Hit@3 accuracy when recommending E3 ligases. Equivariant architectures underperform invariant ones for this prediction, and ESM-2 embeddings improve results only with regularization.
What carries the argument
DegradoMap, a graph neural network that applies lysine-weighted graph pooling with per-protein normalization and cross-attention to capture protein-E3 compatibility.
If this is right
- Degradability and optimal E3 ligase choice can be assessed before any PROTAC molecule is synthesized.
- Well-calibrated outputs with ECE of 0.029 allow experimenters to focus on high-confidence cases first.
- The model supports transfer to new E3 ligases, as shown by the 0.811 AUROC on CRBN-to-VHL evaluation.
- Simpler invariant graph architectures are sufficient and preferable to equivariant designs for this scalar output task.
- Protein language model embeddings require regularization to contribute positively rather than degrade performance.
Where Pith is reading between the lines
- The same lysine-weighted pooling and cross-attention pattern could be adapted to predict efficacy for other proximity-inducing modalities such as molecular glues.
- High seed-to-seed variance implies that deployment would benefit from model ensembles even when average AUROC appears usable.
- Adding additional cellular or tissue-specific context layers beyond the Cancer Dependency Map might further reduce the observed variance.
- For other scalar biophysical prediction tasks, domain-specific pooling may prove more effective than full equivariant message passing.
Load-bearing premise
The PROTAC-8K labels accurately reflect true cellular degradability and performance on the 155 targets and 10 E3 ligases will generalize to proteins and ligases outside this collection.
What would settle it
Measuring actual degradation outcomes for DegradoMap predictions on a set of proteins absent from the 155-target benchmark and comparing those outcomes to the model's probability scores.
Figures








read the original abstract
Proteolysis-targeting chimeras (PROTACs) can selectively degrade disease-causing proteins, yet predicting which targets are amenable to degradation remains a critical bottleneck: existing computational methods require the complete PROTAC molecular structure, information unavailable before synthesis. We present DegradoMap, a graph neural network that predicts PROTAC-mediated degradability from protein structure and E3 ligase identity alone -- the minimal information available at the target selection stage. The model encodes biophysical priors through lysine-weighted graph pooling with per-protein normalization, models protein-E3 compatibility via cross-attention, and integrates cellular context from the Cancer Dependency Map. On the PROTAC-8K benchmark (3,101 samples, 155 targets, 10 E3 ligases), DegradoMap achieves 0.646+-0.124 AUROC on target-unseen evaluation (best seed: 0.7449) and 0.811 AUROC on CRBN->VHL E3-unseen transfer, outperforming GNN and machine learning baselines. The model additionally recommends optimal E3 ligases with 74% Hit@3 accuracy. Two findings carry broader implications: E(3)-equivariant architectures underperform the simpler invariant design for this scalar prediction task, and ESM-2 embeddings improve peak performance only with careful regularization -- naive integration fails. DegradoMap provides pre-synthesis computational guidance for degradability assessment; its well-calibrated confidence scores (ECE = 0.029, target-unseen) enable practitioners to prioritize high-confidence predictions for experimental follow-up. However, the high seed variance (std = 0.124) and limited E3 coverage require ensembling for reliable deployment.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents DegradoMap, a graph neural network that predicts PROTAC-mediated protein degradability using only protein structure and E3 ligase identity (without the PROTAC molecule itself). On the PROTAC-8K benchmark (3101 samples, 155 targets, 10 E3 ligases), it reports 0.646±0.124 AUROC for target-unseen evaluation (best seed 0.7449) and 0.811 AUROC for CRBN-to-VHL E3-unseen transfer, outperforming GNN and ML baselines; it also achieves 74% Hit@3 for E3 ligase recommendation. The model uses lysine-weighted graph pooling, cross-attention for protein-E3 compatibility, and integrates Cancer Dependency Map context. Additional findings note that E(3)-equivariant architectures underperform invariant ones and that naive ESM-2 integration requires regularization.
Significance. If the benchmark labels are reliable, DegradoMap could offer practical pre-synthesis guidance for PROTAC target selection by leveraging minimal input information. The work credits include explicit reporting of high seed-to-seed variance, well-calibrated confidence scores (ECE=0.029), and ablation insights on architecture choices. These elements strengthen the manuscript's utility for practitioners despite the performance numbers.
major comments (3)
- [Abstract] Abstract and (presumed) Methods: The central performance claims (0.646 AUROC target-unseen, 0.811 on E3 transfer) are measured against PROTAC-8K labels, yet no information is given on label provenance, construction method (literature curation, DepMap integration, experimental thresholds), noise estimation, class balance, or orthogonal validation. This is load-bearing because the model receives no PROTAC structure and the labels must accurately reflect true cellular degradability for the AUROCs and baseline comparisons to be interpretable.
- [Abstract] Abstract/Results: The target-unseen AUROC is reported as 0.646 with std=0.124 across seeds (best seed 0.7449), but no methodology is described for computing error bars, number of independent runs, or whether baselines exhibit comparable variance. This undermines assessment of whether the outperformance is statistically reliable given the high variance explicitly noted.
- [Abstract] Abstract: With only 10 E3 ligases in the benchmark, the E3-unseen transfer result (CRBN->VHL at 0.811 AUROC) and the broader claim of generalization to new ligases rest on a narrow set; no analysis of how performance would degrade outside these 10 is provided, which is load-bearing for the transfer claim.
minor comments (2)
- [Abstract] The abstract states 'well-calibrated confidence scores (ECE = 0.029, target-unseen)' but does not define how ECE was computed or on which split; add this detail for reproducibility.
- Notation for the lysine-weighted graph pooling and per-protein normalization should be formalized with equations in the Methods to clarify the biophysical priors.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments, which highlight important aspects of benchmark transparency and statistical rigor. We address each major comment below and indicate where revisions will be made to the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract and (presumed) Methods: The central performance claims (0.646 AUROC target-unseen, 0.811 on E3 transfer) are measured against PROTAC-8K labels, yet no information is given on label provenance, construction method (literature curation, DepMap integration, experimental thresholds), noise estimation, class balance, or orthogonal validation. This is load-bearing because the model receives no PROTAC structure and the labels must accurately reflect true cellular degradability for the AUROCs and baseline comparisons to be interpretable.
Authors: We agree that explicit details on PROTAC-8K label construction are necessary for interpretability. In the revised manuscript we will add a dedicated subsection in Methods describing the benchmark construction, including literature sources, DepMap integration steps, experimental thresholds applied, class balance statistics, and any available estimates of label noise. We will also note the absence of orthogonal validation as a limitation of the current public benchmark. revision: yes
-
Referee: [Abstract] Abstract/Results: The target-unseen AUROC is reported as 0.646 with std=0.124 across seeds (best seed 0.7449), but no methodology is described for computing error bars, number of independent runs, or whether baselines exhibit comparable variance. This undermines assessment of whether the outperformance is statistically reliable given the high variance explicitly noted.
Authors: The reported standard deviation derives from five independent training runs using distinct random seeds. We will clarify this protocol in the revised Methods section and confirm that all baseline models were evaluated under identical conditions (same number of runs and seed strategy) to enable direct comparison of variance. revision: yes
-
Referee: [Abstract] Abstract: With only 10 E3 ligases in the benchmark, the E3-unseen transfer result (CRBN->VHL at 0.811 AUROC) and the broader claim of generalization to new ligases rest on a narrow set; no analysis of how performance would degrade outside these 10 is provided, which is load-bearing for the transfer claim.
Authors: We acknowledge that the E3 ligase coverage is limited to ten ligases and that this constrains strong claims of broad generalization. We will revise the Discussion to explicitly state this limitation and present the CRBN-to-VHL result as a specific transfer case rather than evidence of generalizability beyond the observed set. No further empirical analysis is feasible without additional labeled data. revision: partial
Circularity Check
No circularity: standard supervised GNN evaluation on held-out benchmark splits
full rationale
The paper reports empirical AUROC on target-unseen and E3-unseen splits of the PROTAC-8K benchmark using a GNN that takes protein structure and E3 identity as input. No equations, parameters, or self-citations are presented that reduce the reported performance metrics to fitted values or definitions internal to the model itself. The architecture choices (lysine-weighted pooling, cross-attention, DepMap integration) are described as design decisions rather than derived quantities, and the evaluation follows conventional ML practice with no self-referential prediction steps. The central claims rest on external benchmark labels and standard training, making the result self-contained against the held-out data.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Protein structure plus E3 ligase identity is sufficient to predict degradability without the PROTAC molecule itself.
Reference graph
Works this paper leans on
-
[1]
Békés, D
M. Békés, D. R. Langley, and C. M. Crews. PROTAC targeted protein degraders: the past is prologue.Nature Reviews Drug Discovery, 21(3):181–200, 2022
2022
-
[2]
K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews, and R. J. De- shaies. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation.Proc. Natl. Acad. Sci. USA, 98(15):8554–8559, 2001
2001
-
[3]
Pettersson and C
M. Pettersson and C. M. Crews. PROteolysis TArgeting Chimeras (PROTACs)— past, present and future.Drug Discovery Today: Technologies, 31:15–27, 2019
2019
-
[4]
A. C. Lai and C. M. Crews. Induced protein degradation: an emerging drug discovery paradigm.Nature Reviews Drug Discovery, 16(2):101–114, 2017
2017
-
[5]
F. Li, Q. Hu, X. Zhang, R. Sun, Z. Liu, S. Wu, S. Tian, X. Ma, Z. Dai, X. Yang, S. Gao, and F. Bai. DeepPROTACs is a deep learning-based targeted degradation predictor for PROTACs.Nature Communications, 13:7133, 2022
2022
-
[6]
Ribes, E
S. Ribes, E. Nittinger, C. Tyrchan, and R. Mercado. Modeling PROTAC degradation activity with machine learning.Artificial Intelligence in the Life Sciences, 6:100104, 2024
2024
-
[7]
Zhang, S
W. Zhang, S. S. Roy Burman, J. Chen, K. A. Donovan, Y. Cao, C. Shu, B. Zhang, Z. Zeng, S. Gu, Y. Zhang, D. Li, E. S. Fischer, C. Tokheim, and X. S. Liu. Machine learning modeling of protein-intrinsic features predicts tractability of targeted protein degradation.Genomics, Proteomics & Bioinformatics, 20(5):882–898, 2022
2022
-
[8]
J. Liu, M. J. Roy, L. Isbel, and F. Li. Accurate PROTAC-targeted degradation prediction with DegradeMaster.Bioinformatics, 41(Supplement_1):i342–i351, 2025
2025
-
[9]
Jumper, R
J. Jumper, R. Evans, A. Pritzel, et al. Highly accurate protein structure prediction with AlphaFold.Nature, 596(7873):583–589, 2021
2021
-
[10]
B. Jing, S. Eismann, P. Suriana, R. J. L. Townshend, and R. Dror. Learning from protein structure with geometric vector perceptrons. InProc. ICLR, 2021
2021
-
[11]
Zhang, M
Z. Zhang, M. Xu, A. Jamasb, V. Chenthamarakshan, A. Lozano, P. Das, and J. Tang. Protein representation learning by geometric structure pretraining. InProc. ICLR, 2023
2023
-
[12]
K. T. Schütt, P. J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko, and K. R. Müller. SchNet: a continuous-filter convolutional neural network for modeling quantum interactions. InProc. NeurIPS, pages 991–1001, 2017
2017
-
[13]
V. G. Satorras, E. Hoogeboom, and M. Welling. E(𝑛) equivariant graph neural networks. InProc. ICML, pages 9323–9332, 2021
2021
-
[14]
Z. Lin, H. Akin, R. Rao, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
2023
-
[15]
Elnaggar, M
A. Elnaggar, M. Heinzinger, C. Dallago, et al. ProtTrans: toward understanding the language of life through self-supervised learning.IEEE Trans. PAMI, 44(10):7112– 7127, 2022
2022
-
[16]
C. Hsu, R. Verkuil, J. Liu, Z. Lin, B. Hie, T. Sercu, A. Lerer, and A. Rives. Learning inverse folding from millions of predicted structures. InProc. ICML, 2022
2022
-
[17]
Y. Li, P. Xie, L. Lu, J. Wang, L. Diao, Z. Liu, et al. An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase–substrate interaction network.Nature Communications, 8:347, 2017
2017
-
[18]
H. Fu, Y. Yang, X. Wang, H. Wang, and Y. Xu. DeepUbi: a deep learning framework for prediction of ubiquitination sites in proteins.BMC Bioinformatics, 20:86, 2019
2019
-
[19]
C. Guo, G. Pleiss, Y. Sun, and K. Q. Weinberger. On calibration of modern neural networks. InProc. ICML, pages 1321–1330, 2017
2017
-
[20]
Zengerle, K
M. Zengerle, K. H. Chan, and A. Ciulli. Selective small molecule induced degrada- tion of the BET bromodomain protein BRD4.ACS Chemical Biology, 10(8):1770– 1777, 2015
2015
-
[21]
Tsherniak, F
A. Tsherniak, F. Vazquez, P. G. Montgomery, et al. Defining a cancer dependency map.Cell, 170(3):564–576, 2017
2017
-
[22]
T. Y. Lin, P. Goyal, R. Girshick, K. He, and P. Dollár. Focal loss for dense object detection. InProc. ICCV, pages 2999–3007, 2017
2017
-
[23]
Loshchilov and F
I. Loshchilov and F. Hutter. Decoupled weight decay regularization. InProc. ICLR, 2019
2019
-
[24]
V. Gligorijević, P. D. Renfrew, T. Kosciolek, et al. Structure-based protein function prediction using graph convolutional networks.Nature Communications, 12:3168, 2021. Structure-Aware Prediction of PROTAC-Mediated Protein Degradability via Graph Neural Networks ACM-BCB ’26, June 30 – July 3, 2026, Calabria, Italy Fold 0 Fold 1 Fold 2 Fold 3 Fold 4 Overa...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.