Variational low-energy subspaces for chemically accurate excited states
Pith reviewed 2026-06-28 03:22 UTC · model grok-4.3
The pith
Reformulating excited-state optimization as iterated ground-state problems on low-energy subspaces enables automatic, constraint-free calculation of multiple chemically accurate states.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Variational excited-state optimization reduces to an iterated ground-state-like problem for a low-energy subspace of the electronic Hamiltonian; applying the principle to non-orthogonal Slater determinants produces the automatic EXIDOS method that delivers chemically accurate excited states for a range of molecular systems without symmetry or orthogonality constraints.
What carries the argument
The low-energy subspace of the electronic Hamiltonian, which converts excited-state variational search into repeated ground-state optimizations.
If this is right
- Multiple excited states of different characters are optimized in a single run without prior knowledge of their symmetry or orbital character.
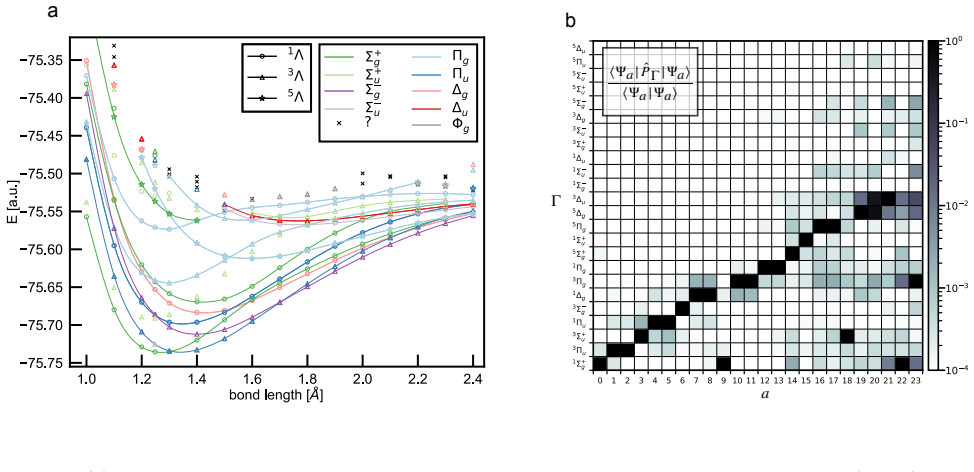
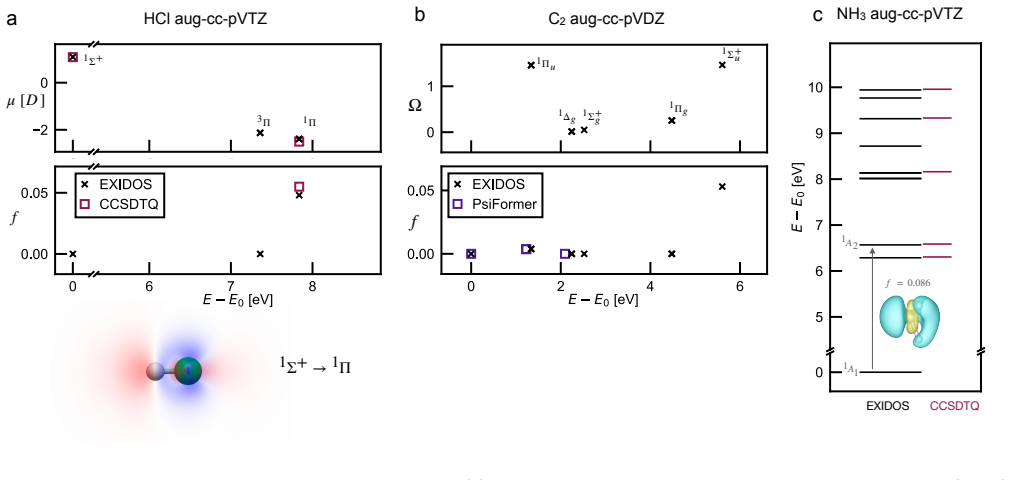
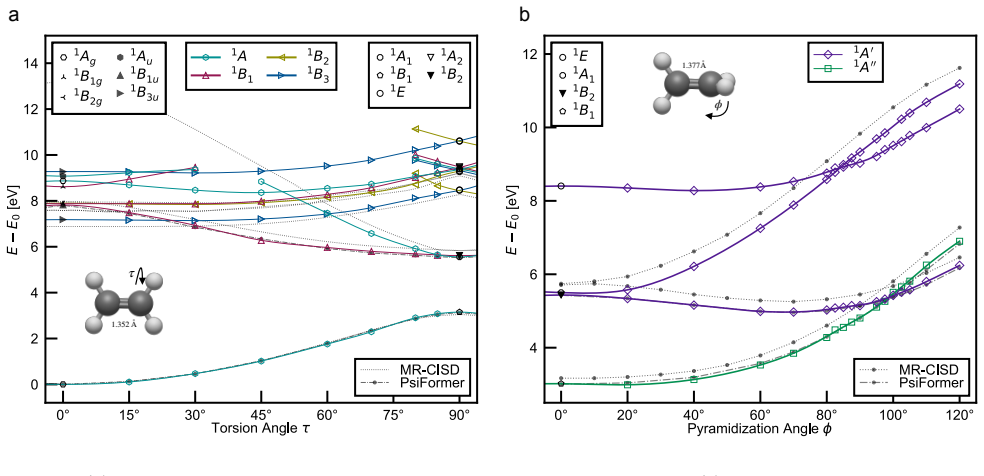
- Chemical accuracy is obtained for charge-transfer states, Rydberg states, double excitations, extended potential-energy curves, avoided crossings and conical intersections.
- The approach remains fully variational and requires only the number of target states and determinants per state as external parameters.
- The method scales more favorably than traditional high-level excited-state techniques while avoiding manual state selection.
Where Pith is reading between the lines
- The automatic character of the procedure may allow routine mapping of entire potential-energy surfaces in photochemical applications where state character changes with geometry.
- Because no orthogonality is enforced by hand, the same framework could be combined with other variational ansatzes that naturally produce non-orthogonal states.
- The subspace construction may generalize to larger active spaces or embedding schemes once the determinant optimization step is replaced by a more scalable solver.
Load-bearing premise
The reformulation of excited-state optimization as iterated ground-state problems on low-energy subspaces remains valid when the trial functions are non-orthogonal Slater determinants that carry no explicit orthogonality or symmetry constraints.
What would settle it
A full-configuration-interaction benchmark on any of the tested molecules in which EXIDOS energies for one or more states deviate by more than chemical accuracy (approximately 1 kcal/mol) when run with the stated number of states and determinants and without manual symmetry input.
Figures

read the original abstract
Accurate electronic excited states are essential for photochemistry, spectroscopy and non-adiabatic molecular dynamics, but high-level calculations often scale steeply and require prior knowledge of the target state's character or symmetry. Here we show that variational excited-state optimization can be reformulated as an iterated ground-state-like problem for a low-energy subspace of the electronic Hamiltonian. Applying this variational principle to non-orthogonal Slater determinants leads to EXIDOS, an automatic method for excited state calculations controlled only by the number of states and determinants per state. EXIDOS optimizes multiple excited states simultaneously, without explicit orthogonality constraints or imposed spin and point-group symmetries. Benchmarks against FCI and state-of-the-art quantum chemistry methods show chemical accuracy for a multitude of states in N$_2$ and CO, charge-transfer states in HCl, Rydberg states in NH$_3$, double excitations and extended potential-energy curves in C$_2$, and avoided crossings and conical intersections in ethylene. These results establish EXIDOS as a low-scaling, fully variational route to chemically accurate excited states.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents EXIDOS, a method reformulating variational excited-state optimization as an iterated ground-state-like problem over a low-energy subspace of the electronic Hamiltonian. Applied to non-orthogonal Slater determinants without explicit orthogonality, spin, or point-group symmetry constraints, it claims to achieve chemical accuracy for multiple states in N2 and CO, charge-transfer states in HCl, Rydberg states in NH3, double excitations and potential-energy curves in C2, and avoided crossings/conical intersections in ethylene, as shown in benchmarks against FCI and other quantum chemistry methods.
Significance. If the reformulation is variationally sound, this constitutes a notable advance as a fully variational, low-scaling route to chemically accurate excited states that requires only the number of states and determinants per state as input, eliminating the need for prior state character or symmetry knowledge. The breadth of benchmark systems (including challenging cases like double excitations and conical intersections) would support broad applicability in photochemistry and non-adiabatic dynamics if the underlying principle holds without artifacts from the optimization procedure.
major comments (2)
- [Abstract and variational reformulation section] The central reformulation (abstract and the section deriving the subspace variational principle): no explicit derivation or proof is supplied showing that the low-energy subspace energy functional remains bounded, unique, and automatically enforces state separation when trial functions are non-orthogonal Slater determinants without imposed orthogonality constraints. This is load-bearing for the claim that chemical accuracy on ethylene conical intersections and C2 double excitations follows from the variational principle rather than procedure-specific details.
- [Benchmark results section] Benchmark section (results on N2, C2, ethylene): the reported chemical accuracy is asserted without accompanying discussion of convergence criteria, implementation of the iterated subspace optimization, or analysis of possible failure modes (e.g., mixing or collapse) under the relaxed symmetry conditions. This undermines assessment of whether the results generalize beyond the tested cases.
minor comments (2)
- [Abstract] The acronym EXIDOS is used in the abstract without expansion or definition; introduce it with its full meaning on first use.
- [References] Ensure all compared state-of-the-art methods (e.g., those benchmarked against FCI) have complete and up-to-date citations in the reference list.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the positive assessment of its potential significance. We respond to the two major comments below and will incorporate revisions as indicated.
read point-by-point responses
-
Referee: [Abstract and variational reformulation section] The central reformulation (abstract and the section deriving the subspace variational principle): no explicit derivation or proof is supplied showing that the low-energy subspace energy functional remains bounded, unique, and automatically enforces state separation when trial functions are non-orthogonal Slater determinants without imposed orthogonality constraints. This is load-bearing for the claim that chemical accuracy on ethylene conical intersections and C2 double excitations follows from the variational principle rather than procedure-specific details.
Authors: We agree that the manuscript presents the subspace variational principle at a conceptual level without a self-contained mathematical derivation or proof of boundedness, uniqueness, and automatic state separation for non-orthogonal Slater determinants. This omission weakens the link between the variational principle and the reported accuracies. In the revised manuscript we will add an explicit derivation (as a new subsection or appendix) that proves the low-energy subspace functional is bounded from below by the sum of the lowest eigenvalues, is unique under the stated conditions, and enforces separation without explicit orthogonality constraints. This addition will demonstrate that the chemical accuracy on the cited systems follows directly from the variational principle. revision: yes
-
Referee: [Benchmark results section] Benchmark section (results on N2, C2, ethylene): the reported chemical accuracy is asserted without accompanying discussion of convergence criteria, implementation of the iterated subspace optimization, or analysis of possible failure modes (e.g., mixing or collapse) under the relaxed symmetry conditions. This undermines assessment of whether the results generalize beyond the tested cases.
Authors: We concur that the benchmark section lacks sufficient detail on convergence criteria, the precise implementation of the iterated subspace optimization, and analysis of failure modes such as mixing or collapse when symmetry constraints are relaxed. These omissions limit evaluation of robustness and generalizability. In the revised manuscript we will expand the benchmark section to include: explicit convergence thresholds and stopping criteria; a step-by-step description of the iterated optimization procedure; and a dedicated analysis of potential failure modes, supported by additional numerical diagnostics on the N2, C2, and ethylene systems showing that mixing or collapse did not occur. revision: yes
Circularity Check
No significant circularity; reformulation is presented as independent variational principle
full rationale
The paper introduces a reformulation of excited-state optimization as an iterated ground-state problem on a low-energy subspace, then applies it to non-orthogonal Slater determinants to define EXIDOS. The abstract and description frame this as a new variational principle controlled only by number of states and determinants, with external benchmarks against FCI and other methods. No equations or steps are shown that reduce the claimed accuracy or the subspace minimum to fitted inputs, self-definitions, or self-citation chains by construction. The central claim rests on the validity of the reformulation itself rather than on any renaming or imported uniqueness theorem from the authors' prior work.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Variational excited-state optimization can be reformulated as an iterated ground-state-like problem for a low-energy subspace of the electronic Hamiltonian.
Reference graph
Works this paper leans on
-
[1]
Casida and M
M. Casida and M. Huix-Rotllant, Progress in time- dependent density-functional theory, Annual Review of Physical Chemistry63, 287–323 (2012)
2012
-
[2]
Dreuw and M
A. Dreuw and M. Head-Gordon, Single-reference ab ini- tio methods for the calculation of excited states of large molecules, Chemical Reviews105, 4009–4037 (2005)
2005
-
[3]
Dreuw and M
A. Dreuw and M. Wormit, The algebraic diagrammatic construction scheme for the polarization propagator for the calculation of excited states, WIREs Computational Molecular Science5, 82–95 (2014)
2014
-
[4]
H.KochandP.Jørgensen,Coupledclusterresponsefunc- tions, The Journal of Chemical Physics93, 3333–3344 (1990)
1990
-
[5]
Christiansen, H
O. Christiansen, H. Koch, and P. Jo/rgensen, Response functions in the cc3 iterative triple excitation model, The Journal of Chemical Physics103, 7429–7441 (1995)
1995
-
[6]
J. F. Stanton and R. J. Bartlett, The equation of motion coupled-cluster method. a systematic biorthogonal ap- proach to molecular excitation energies, transition prob- abilities, and excited state properties, The Journal of Chemical Physics98, 7029–7039 (1993)
1993
-
[7]
A. I. Krylov, Equation-of-motion coupled-cluster meth- ods for open-shell and electronically excited species: The hitchhiker’s guide to fock space, Annual Review of Phys- ical Chemistry59, 433 (2008)
2008
-
[8]
W. Purwanto, S. Zhang, and H. Krakauer, Excited state calculations using phaseless auxiliary-field quan- tum monte carlo: Potential energy curves of low-lying c2 singlet states, The Journal of Chemical Physics130, 10.1063/1.3077920 (2009)
-
[9]
Sneskov and O
K. Sneskov and O. Christiansen, Excited state coupled cluster methods, WIREs Computational Molecular Sci- ence2, 566–584 (2011)
2011
-
[10]
Lischka, D
H. Lischka, D. Nachtigallová, A. J. A. Aquino, P. G. Szalay, F. Plasser, F. B. C. Machado, and M. Barbatti, Multireference approaches for excited states of molecules, Chemical Reviews118, 7293 (2018)
2018
-
[11]
Serrano-Andrés, M
L. Serrano-Andrés, M. Merchán, I. Nebot-Gil, R. Lindh, and B. O. Roos, Towards an accurate molecular orbital theory for excited states: Ethene, butadiene, and hexa- triene, The Journal of Chemical Physics98, 3151–3162 (1993)
1993
-
[12]
Angeli, R
C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger, and J.-P. Malrieu, Introduction of n-electron valence states for multireference perturbation theory, The Jour- nal of Chemical Physics114, 10252 (2001)
2001
-
[13]
A. A. Holmes, C. J. Umrigar, and S. Sharma, Ex- cited states using semistochastic heat-bath configura- tion interaction, The Journal of Chemical Physics147, 10.1063/1.4998614 (2017)
-
[14]
N. S. Blunt, S. D. Smart, G. H. Booth, and A. Alavi, An excited-state approach within full configuration in- teraction quantum monte carlo, The Journal of Chemical Physics143, 10.1063/1.4932595 (2015)
-
[15]
C. A. Jiménez-Hoyos, R. Rodríguez-Guzmán, and G. E. Scuseria, Excited electronic states from a variational approach based on symmetry-projected hartree–fock configurations, The Journal of Chemical Physics139, 10.1063/1.4840097 (2013)
-
[16]
Nite and C
J. Nite and C. A. Jiménez-Hoyos, Low-cost molec- ular excited states from a state-averaged resonating hartree–fock approach, Journal of Chemical Theory and Computation15, 5343–5351 (2019)
2019
-
[17]
D. Pfau, S. Axelrod, H. Sutterud, I. von Glehn, and J. S. Spencer, Accurate computation of quantum excited states with neural networks, Science385, 10.1126/sci- ence.adn0137 (2024)
-
[18]
Y. Ma, C. Liu, W. Li, S.-Y. Zhang, L. M. Duan, Y. Wu, and D.-L. Deng, Solving excited states for long-range interacting trapped ions with neural networks (2025), arXiv:2506.08594
arXiv 2025
- [19]
-
[20]
A. Kahn, L. Gravina, and F. Vicentini, Variational sub- space methods and application to improving variational monte carlo dynamics, Quantum10, 2082 (2026)
2082
-
[21]
S.-X. Zhang and L. Wang, A unified variational framework for quantum excited states (2025), arXiv:2504.21459
arXiv 2025
-
[22]
C. Giuliani, J. Nys, R. Martinazzo, G. Carleo, and R. Rossi, Precise quantum chemistry calculations with few slater determinants, Nature Communications 10.1038/s41467-026-70255-z (2026)
-
[23]
P.-F. Loos, A. Scemama, A. Blondel, Y. Garniron, M. Caffarel, and D. Jacquemin, A mountaineering strat- egy to excited states: Highly accurate reference energies and benchmarks, Journal of Chemical Theory and Com- putation14, 4360–4379 (2018)
2018
-
[24]
P.-F. Loos, M. Boggio-Pasqua, A. Blondel, F. Lipparini, and D. Jacquemin, Quest database of highly-accurate ex- citation energies, Journal of Chemical Theory and Com- putation21, 8010–8033 (2025)
2025
-
[25]
G. E. Scuseria, C. A. Jiménez-Hoyos, T. M. Henderson, K. Samanta, and J. K. Ellis, Projected quasiparticle the- ory for molecular electronic structure, The Journal of Chemical Physics135, 10.1063/1.3643338 (2011)
-
[26]
C. A. Jiménez-Hoyos, T. M. Henderson, T. Tsuchimochi, and G. E. Scuseria, Projected hartree–fock theory, The Journal of Chemical Physics136, 10.1063/1.4705280 (2012)
-
[27]
Chrayteh, A
A. Chrayteh, A. Blondel, P.-F. Loos, and D. Jacquemin, Mountaineering strategy to excited states: Highly ac- curate oscillator strengths and dipole moments of small molecules, Journal of Chemical Theory and Computation 17, 416–438 (2020)
2020
-
[28]
M. T. do Casal, J. M. Toldo, M. Barbatti, and F. Plasser, Classification of doubly excited molecular electronic states, Chemical Science14, 4012–4026 (2023)
2023
-
[29]
Barbatti, J
M. Barbatti, J. Paier, and H. Lischka, Photochemistry of ethylene: A multireference configuration interaction investigation of the excited-state energy surfaces, The Journal of Chemical Physics121, 11614–11624 (2004). 11
2004
-
[30]
S. Sharma, A general non-abelian density matrix renor- malization group algorithm with application to the c2 dimer, The Journal of Chemical Physics142, 10.1063/1.4905237 (2015)
-
[31]
P.-F. Loos, M. Boggio-Pasqua, A. Scemama, M. Caffarel, and D. Jacquemin, Reference energies for double excita- tions, Journal of Chemical Theory and Computation15, 1939–1956 (2019)
1939
-
[32]
G. Levi, M. Kroesbergen, L. Thirion, Y. L. A. Schmer- witz, E. Ö. Jónsson, P. Bilous, P. Hansmann, and H. Jónsson, Orbital optimization and neural-network- assisted configuration interaction calculations of rydberg states, Journal of Chemical Theory and Computation22, 3260–3267 (2026)
2026
-
[33]
D. Feller, K. A. Peterson, and E. R. Davidson, A system- aticapproachtoverticallyexcitedstatesofethyleneusing configuration interaction and coupled cluster techniques, TheJournal ofChemical Physics141,10.1063/1.4894482 (2014)
-
[34]
Y. L. A. Schmerwitz, A. V. Ivanov, E. O. Jónsson, H. Jónsson, and G. Levi, Variational density functional calculations of excited states: Conical intersection and avoided crossing in ethylene bond twisting, The Journal of Physical Chemistry Letters13, 3990–3999 (2022)
2022
-
[35]
M. T. Entwistle, Z. Schätzle, P. A. Erdman, J. Her- mann, and F. Noé, Electronic excited states in deep variational monte carlo, Nature Communications14, 10.1038/s41467-022-35534-5 (2023)
-
[36]
Saade and H
S. Saade and H. G. A. Burton, Excited state-specific casscftheoryforthetorsionofethylene,JournalofChem- ical Theory and Computation20, 5105–5114 (2024)
2024
-
[37]
S. D. Pineda Flores and E. Neuscamman, Excited state specific multi-slater jastrow wave functions, The Journal of Physical Chemistry A123, 1487 (2019)
2019
-
[38]
Schautz and C
F. Schautz and C. Filippi, Optimized jastrow–slater wave functions for ground and excited states: Application to the lowest states of ethene, The Journal of Chemical Physics120, 10931–10941 (2004)
2004
-
[39]
H. G. A. Burton and A. J. W. Thom, Reaching full correlation through nonorthogonal configuration interaction: A second-order perturbative approach, Journal of Chemical Theory and Computation16, 10.1021/acs.jctc.0c00468 (2020)
-
[40]
A. Mahajan, S. Sharma, S. Zhang, and D. R. Reichman, Excited states in auxiliary field quantum monte carlo, TheJournal ofChemical Physics164,10.1063/5.0302374 (2026)
-
[41]
Zhang, H
J. Zhang, H. Song, and Y. Liu, Permanent variational wavefunctionsforbosons,PhysicaA:StatisticalMechan- ics and its Applications599, 127399 (2022)
2022
-
[42]
Tr” for the operator trace, and the normal-case symbol “tr
B. Que, J. Zhang, H. Song, and Y. Liu, Hartree–fock approximation for bosons with symmetry-adapted vari- ational wave functions, Physica A: Statistical Mechanics and its Applications664, 130449 (2025). 12 Appendix Appendix A: Derivation of the effective operators LetU={|Ψ a⟩}K−1 a=0 be a set of variational states drawn from independent variational manifol...
2025
-
[43]
Then we Cholesky- decompose ˜S=LL † and use Lanczos to find the lowest eigenvector ofC=L−1 ˜H(L†)−1, transforming back at the end withL
Projecting out the nullspace and solving the generalized eigenvalue problem We numerically construct the projection matrix by diagonalizing eachm×mblock on the diagonal of ˜S. Then we Cholesky- decompose ˜S=LL † and use Lanczos to find the lowest eigenvector ofC=L−1 ˜H(L†)−1, transforming back at the end withL. For Lanczos solver we can use the previous c...
-
[44]
Numerical stability To ensure numerical stability we normalize all states|Ψa⟩, and for the calculation of the matrix elements we also normalize the orbitals of the individual determinants|Φik⟩and use a sufficiently large Krylov space to render the Lanczos solver numerically stable. Appendix D: Dipole moment and oscillator strentgh We compute the dipole mo...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.