The Metric Picks the Winner: Evaluation Choice Flips Model Rankings for Drug-Response Prediction in Unseen Chemistry
Pith reviewed 2026-06-27 10:24 UTC · model grok-4.3
The pith
The choice of evaluation metric reverses which models win at predicting drug responses on unseen compounds.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
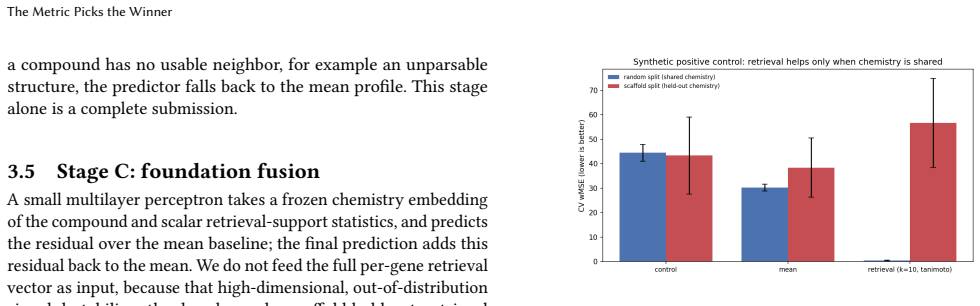
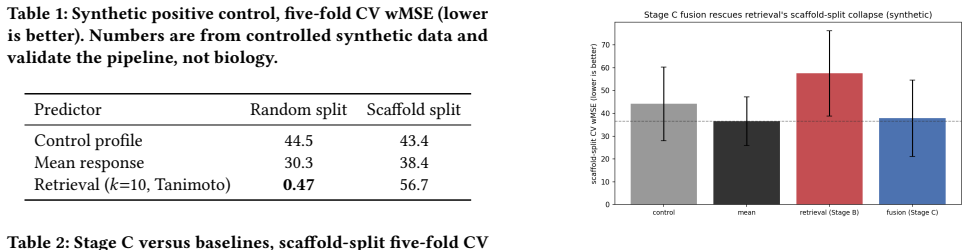
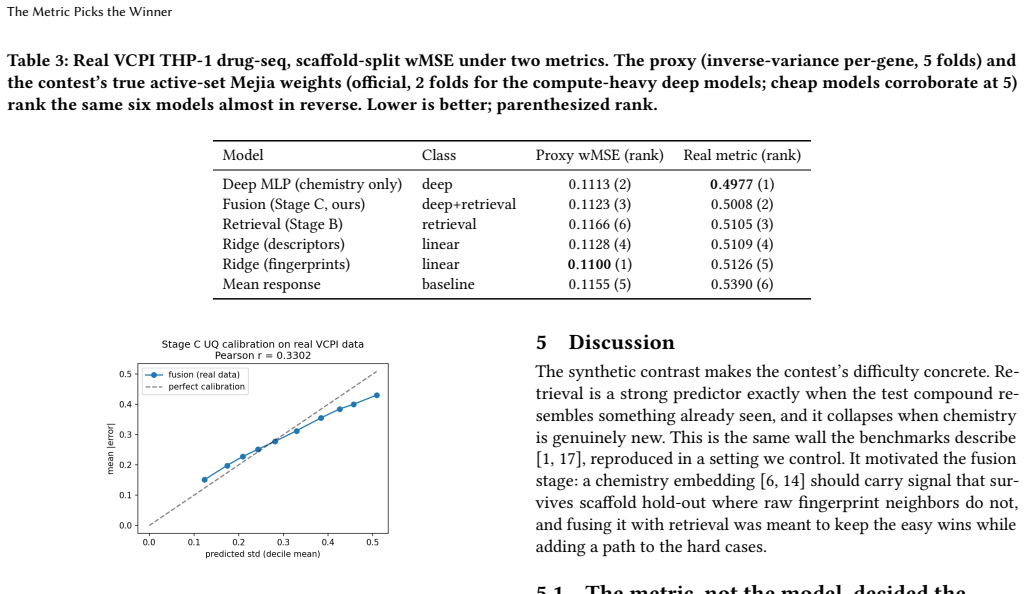
On the VCPI THP-1 DRUG-seq data under a Bemis-Murcko scaffold split, the model ranking flips with metric: the inverse-variance per-gene proxy ranks regularized linear regression on Morgan fingerprints highest, while the contest's per-(gene, compound) Mejia-weighted MSE ranks the deep fusion decoder highest, beating the linear baseline by 0.012 wMSE with paired bootstrap p < 10^-4; the proxy's apparent winner becomes the worst chemistry-aware predictor under the official scoring rule.
What carries the argument
The active-set weighted MSE (wMSE) that applies per-(gene, compound) Mejia weights to the 1064 x 12995 prediction grid, contrasted with an inverse-variance per-gene proxy, on data split by Bemis-Murcko scaffolds.
If this is right
- Deep models and retrieval-augmented fusion outperform linear fingerprint baselines once the evaluation uses the contest's active-set weights.
- A frozen chemistry embedding plus retrieval-support features can predict residuals over the mean response with an added uncertainty head.
- Proxy metrics calibrated only on variance per gene can invert the ordering of chemistry-aware predictors on held-out scaffolds.
- Releasing a pipeline that emits valid submissions directly to the official scorer allows direct comparison against the organizers' 0.507 reference.
Where Pith is reading between the lines
- Benchmarks in perturbation prediction should default to the target contest metric rather than variance proxies to avoid selecting models that will underperform in deployment.
- The same metric-sensitivity pattern observed here for drug chemistry may appear when other held-out axes (dose, time, cell type) are used.
- An uncertainty head trained alongside the residual predictor could be used to flag compounds whose predictions carry high risk under the weighted metric.
Load-bearing premise
The Bemis-Murcko scaffold split on the THP-1 data produces a test chemistry distribution that matches the intended use case of truly unseen compounds.
What would settle it
Re-evaluating the same set of submitted predictions on the official VCPI scorer and finding that the linear fingerprint model still outranks the fusion decoder under the Mejia weights.
Figures
read the original abstract
Predicting how a cell's transcriptome responds to a drug it has never seen is a core, hard problem in computational cell biology: recent benchmarks show complex models often fail to beat trivial baselines once test compounds are held out by chemistry. We study one cell line and assay, THP-1 cells profiled by DRUG-seq, scored by the active-compound weighted MSE(wMSE) of the VCPI prediction contest. We propose a staged approach: dumb baselines (untreated control and mean training-compound response) that the field keeps failing to beat; non-parametric retrieval (a Tanimoto-weighted average of a held-out compound's nearest training compounds); and a fusion stage combining a frozen chemistry embedding with retrieval-support features to predict the residual over the mean, with an uncertainty head and gene programs. On the released VCPI THP-1 drug-seq data (14,026 training compounds), under a Bemis-Murcko scaffold split, the model ranking inverts depending on the metric. Under an inverse-variance per-gene proxy, a regularized linear regression on Morgan fingerprints appears to win over the deep models, retrieval, and ChemBERTa -- the textbook "simple baselines win" result. But under the contest's true active-set metric (per-(gene, compound) Mejia weights, validated against the official scorer; mean baseline 0.535 vs the organizers' 0.507 reference), that reverses: the deep models win, our fusion decoder significantly beats the linear fingerprint baseline (-0.012 wMSE, paired bootstrap p < 10^-4), and the proxy's winner becomes the worst chemistry-aware predictor. Picking the metric picks the winner -- to our knowledge the first demonstration on real held-out drug chemistry of the metric-calibration effect established largely on genetic perturbation. We release a reproducible pipeline wired to the official scorer that emits a valid submission over the real 1064 x 12,995 grid.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that in drug-response prediction on THP-1 DRUG-seq data under a Bemis-Murcko scaffold split, evaluation metric choice inverts model rankings: an inverse-variance per-gene proxy favors regularized linear Morgan fingerprint regression over deep models and retrieval, while the contest's true active-set wMSE (per-(gene, compound) Mejia weights, validated on the official scorer) reverses this, with the authors' fusion decoder (frozen chemistry embedding + retrieval features + uncertainty head + gene programs) beating the linear baseline by -0.012 wMSE (paired bootstrap p < 10^-4). Dumb baselines and non-parametric retrieval are also evaluated, and a reproducible pipeline emitting valid submissions on the 1064 x 12,995 grid is released.
Significance. If the result holds, it provides the first demonstration on real held-out drug chemistry of the metric-calibration effect previously noted mainly on genetic perturbations, showing that proxy metrics can produce misleading rankings in this domain. The release of a reproducible pipeline wired to the official scorer is a clear strength that supports verification and reuse.
major comments (1)
- [Abstract] Abstract: The central claim of a demonstration 'on real held-out drug chemistry' rests on the Bemis-Murcko scaffold split producing a test chemistry distribution that matches the intended use case of truly unseen compounds. No quantitative validation of this assumption (e.g., pairwise Tanimoto histograms between train/test sets, scaffold overlap statistics, or comparison against temporal/external splits) is reported, leaving open the possibility that residual chemical similarity drives the observed inversion rather than a robust metric property.
Simulated Author's Rebuttal
We thank the referee for the constructive comment on validating the chemical novelty of the Bemis-Murcko split. We address it directly below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim of a demonstration 'on real held-out drug chemistry' rests on the Bemis-Murcko scaffold split producing a test chemistry distribution that matches the intended use case of truly unseen compounds. No quantitative validation of this assumption (e.g., pairwise Tanimoto histograms between train/test sets, scaffold overlap statistics, or comparison against temporal/external splits) is reported, leaving open the possibility that residual chemical similarity drives the observed inversion rather than a robust metric property.
Authors: We agree that explicit quantitative support for the degree of chemical novelty would strengthen the interpretation. In revision we will add (i) histograms of per-test-compound maximum Tanimoto similarity to the training set and (ii) scaffold-overlap statistics. Bemis-Murcko splitting remains the community-standard protocol for enforcing chemical hold-out in this domain; the metric-inversion result is demonstrated under that protocol. However, the released THP-1 DRUG-seq dataset contains no temporal metadata, so a temporal-split comparison cannot be performed. revision: partial
- Comparison against temporal or external splits, because the dataset provides no temporal information.
Circularity Check
No circularity: empirical ranking inversion on external contest metric
full rationale
The paper reports an empirical comparison of models on a fixed Bemis-Murcko scaffold split of the VCPI THP-1 dataset, evaluating wMSE under two different weighting schemes (inverse-variance proxy vs. the contest's per-(gene,compound) Mejia weights). The reported result (-0.012 wMSE advantage for the fusion decoder under Mejia weights, with bootstrap p-value) is a direct computation against an externally defined scorer and standard baselines; no equations, fitted parameters, or self-citations reduce this difference to a quantity defined by the authors' own inputs. The derivation chain consists of standard training, inference, and metric application steps that remain independent of the target claim.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Bemis-Murcko scaffold split produces a representative unseen-chemistry test distribution
- domain assumption The contest wMSE (Mejia weights) is the appropriate ground-truth metric for model selection
Reference graph
Works this paper leans on
-
[1]
Constantin Ahlmann-Eltze, Wolfgang Huber, and Simon Anders. 2025. Deep- learning-based gene perturbation effect prediction does not yet outperform simple linear baselines.Nature Methods(2025). https://www.nature.com/articles/ s41592-025-02772-6
2025
-
[2]
Walid Ahmad, Elana Simon, Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. 2022. ChemBERTa-2: Towards Chemical Foundation Models. arXiv:2209.01712. https://arxiv.org/abs/2209.01712
arXiv 2022
-
[3]
Gerold Csendes, Gema Sanz, Kristóf Z. Szalay, and Bence Szalai. 2025. Bench- marking foundation cell models for post-perturbation RNA-seq prediction. BMC Genomics 26:412 (2025). https://pubmed.ncbi.nlm.nih.gov/40269681/
arXiv 2025
-
[4]
Samuel G. Finlayson, Matthew B. A. McDermott, Alex V. Pickering, Scott L. Lipnick, and Isaac S. Kohane. 2019. Cross-modal representation alignment of molecular structure and perturbation-induced transcriptional profiles. arXiv. https://arxiv.org/pdf/1911.10241
arXiv 2019
-
[5]
Mahshid Heidari, Mina Karimpour, Sumana Srivatsa, and Hesam Montazeri. 2026. Evaluating Single-Cell Perturbation Response Models Is Far from Straightforward. bioRxiv. https://www.biorxiv.org/content/10.64898/2026.02.14.705879v1.full 6 The Metric Picks the Winner
-
[6]
Leon Hetzel et al. 2022. Predicting Cellular Responses to Novel Drug Pertur- bations at a Single-Cell Level (chemCPA). InNeurIPS. https://proceedings. neurips.cc/paper_files/paper/2022/file/aa933b5abc1be30baece1d230ec575a7- Paper-Conference.pdf
2022
-
[7]
Yiming Li, Min Zeng, Jun Zhu, Linjing Liu, Fang Wang, Longkai Huang, Fan Yang, Min Li, and Jianhua Yao. 2025. Genetic-to-Chemical Perturbation Transfer Learning Through Unified Multimodal Molecular Representations. bioRxiv. https://www.biorxiv.org/content/10.1101/2025.02.02.635055.full.pdf
-
[8]
Mohammad Lotfollahi et al . 2023. Predicting cellular responses to complex perturbations in high-throughput screens (CPA).Molecular Systems Biology (2023). https://www.embopress.org/doi/abs/10.15252/msb.202211517
-
[9]
Gabriel M. Mejia, Henry E. Miller, Francis J. A. Leblanc, Bo Wang, Brendan Swain, and Lucas Paulo de Lima Camillo. 2025. Diversity by Design: Addressing Mode Collapse Improves scRNA-seq Perturbation Modeling on Well-Calibrated Metrics. arXiv:2506.22641. https://arxiv.org/abs/2506.22641
arXiv 2025
-
[10]
Oscar Méndez-Lucio, Christos Nicolaou, and Berton Earnshaw. 2024. MolE: a molecular foundation model for drug discovery. arXiv / Nature Communications. https://arxiv.org/pdf/2211.02657
arXiv 2024
-
[11]
Fanbo Meng, Can Wang, Yue Lin, Jing Mo, Xunzhi Zhang, Zhaotong Cong, Chi Song, Sanyin Zhang, Shilin Chen, Liang Leng, and Wei Chen. 2026. DeepICER: A deep learning framework for predicting compound-induced gene expression profiles.Acta Pharmaceutica Sinica B(2026). https://www.sciencedirect.com/ science/article/pii/S2211383526001000
2026
-
[12]
Miller, Gabriel M
Henry E. Miller, Gabriel M. Mejia, Francis J. A. Leblanc, Bo Wang, Brendan Swain, and Lucas Paulo de Lima Camillo. 2025. Deep Learning-Based Genetic Perturbation Models Do Outperform Uninformative Baselines on Well-Calibrated Metrics. bioRxiv 2025.10.20.683304. https://www.biorxiv.org/content/10.1101/ 2025.10.20.683304v1
2025
-
[13]
Xiaoning Qi et al. 2024. Predicting transcriptional responses to novel chemical perturbations using a deep generative model for drug discovery (PRnet).Nature Communications(2024). https://www.nature.com/articles/s41467-024-53457-1
2024
-
[14]
Eduardo Soares, Victor Shirasuna, Emilio Vital Brazil, Renato Cerqueira, Dmitry Zubarev, and Kristin Schmidt. 2024. A Large Encoder-Decoder Family of Foun- dation Models for Chemical Language. arXiv. https://arxiv.org/pdf/2407.20267
arXiv 2024
-
[15]
Xiaochu Tong, Ning Qu, Xiangtai Kong, Shengkun Ni, Jingyi Zhou, Kun Wang, Lehan Zhang, Yiming Wen, Jiangshan Shi, Sulin Zhang, Xutong Li, and Mingyue Zheng. 2023. TranSiGen: Deep representation learning of chemical-induced transcriptional profile. bioRxiv. https://www.biorxiv.org/content/10.1101/2023. 11.12.566777.full.pdf
-
[16]
Augustin Wenteler, Martina Occhetta, Nikhil Branson, Magdalena Huebner, Vic- tor Curean, William T. Dee, William T. Connell, Alex Hawkins-Hooker, Si Pham Chung, Yasha Ektefaie, Aoife Gallagher-Syed, and Charlotte M. V. Córdova. 2024. PertEval-scFM: Benchmarking Single-Cell Foundation Models for Perturbation Effect Prediction. bioRxiv. https://www.biorxiv....
-
[17]
Yan Wu, Esther Wershof, Sebastian M. Schmon, Marcel Nassar, Błażej Osiński, Ridvan Eksi, Zichao Yan, Rory Stark, Kun Zhang, and Thore Graepel. 2024. PerturBench: Benchmarking Machine Learning Models for Cellular Perturbation Analysis. arXiv. https://arxiv.org/html/2408.10609v4
arXiv 2024
-
[18]
Chaoyang Ye et al . 2018. DRUG-seq for miniaturized high-throughput tran- scriptome profiling in drug discovery.Nature Communications(2018). https: //pmc.ncbi.nlm.nih.gov/articles/PMC6192987/
2018
-
[19]
Linchang Zhu and Yuanhanyu Luo. 2025. DrugPT: A Flexible Framework for In- tegrating Gene and Chemical Representations in Perturbation Modeling. bioRxiv. https://www.biorxiv.org/content/10.1101/2025.07.25.665130.full.pdf 7
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.