scLLM-DSC: LLM-Knowledge Enhanced Cross-Modal Deep Structural Clustering for Single-Cell RNA Sequencing
Pith reviewed 2026-06-27 07:41 UTC · model grok-4.3
The pith
scLLM-DSC aligns LLM-derived gene semantics with transcriptomic features via cross-modal contrastive learning to improve single-cell RNA clustering.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
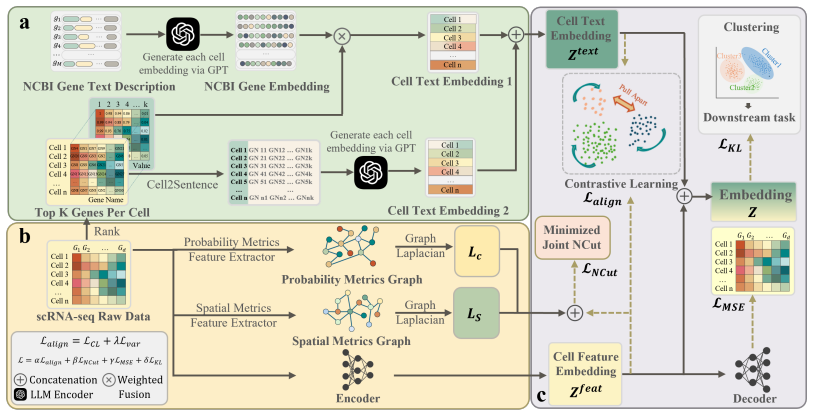
scLLM-DSC creates a semantically grounded representation by combining a Knowledge-Driven Semantic View derived from NCBI gene priors and contextualized Cell2Sentence embeddings with a Structure-Aware Topological View extracted via a graph-guided encoder, then applies cross-modal contrastive alignment to enforce consistency between biological semantics and transcriptomic features inside a unified latent space.
What carries the argument
The cross-modal contrastive alignment mechanism that enforces consistency between the knowledge-driven semantic view and the structure-aware topological view.
If this is right
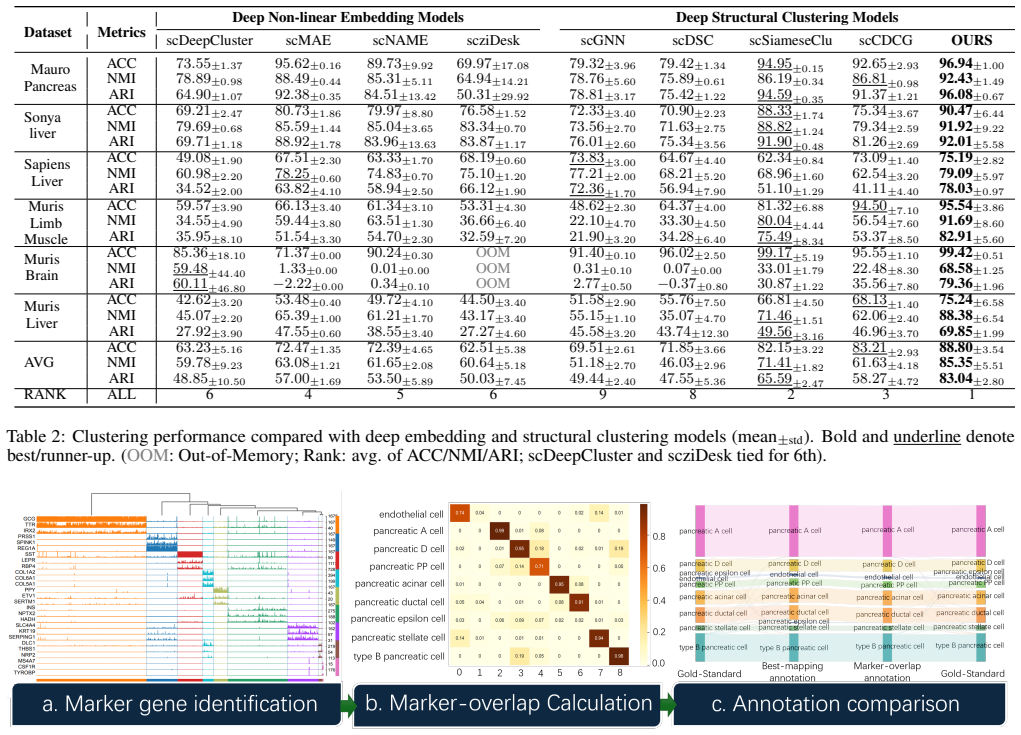
- Clustering accuracy exceeds that of eleven existing state-of-the-art methods on benchmark datasets.
- Cell populations are identified with representations that incorporate both numerical structure and explicit biological gene function.
- Tissue heterogeneity is resolved more effectively because the latent space respects semantic consistency across modalities.
- The same alignment procedure can be applied to new datasets without retraining the underlying LLM components from scratch.
- Downstream tasks that depend on accurate cell-type separation become more reliable once the unified representation is obtained.
Where Pith is reading between the lines
- The contrastive step may allow other generative models besides the specific Cell2Sentence setup to supply useful priors for single-cell tasks.
- If the alignment generalizes, similar cross-modal techniques could be tested on multi-omics datasets where one modality already carries functional annotations.
- Removing reliance on any single knowledge base such as NCBI would test whether the performance lift is robust to different sources of gene semantics.
- The framework implies that future work could measure how much of the accuracy gain comes from the topological encoder versus the semantic view alone.
Load-bearing premise
Biological semantics extracted from NCBI gene priors and Cell2Sentence embeddings can be meaningfully aligned with raw transcriptomic features via contrastive learning in a way that improves downstream clustering.
What would settle it
An experiment on standard scRNA-seq datasets in which scLLM-DSC shows no clustering accuracy gain over the eleven state-of-the-art baselines when the contrastive alignment step is removed or when the semantic view is replaced by random embeddings.
Figures



read the original abstract
Clustering is fundamental to scRNA-seq analysis, serving as a cornerstone for identifying cell populations and resolving tissue heterogeneity. However, existing methods focus on mining numerical statistical patterns, suffering from semantic agnosticism by neglecting the intrinsic biological functions encoded by genes. While Large Language Models (LLMs) offer promising semantic capabilities, their direct adaptation to cell clustering is hindered by the structural mismatch between generative pre-training objectives and discriminative downstream tasks. To bridge this gap, we propose scLLM-DSC, a novel LLM-Knowledge Enhanced Cross-Modal Deep Structural Clustering framework. Diverging from data-driven paradigms, scLLM-DSC establishes a semantically-grounded representation by synergizing two views: a Knowledge-Driven Semantic View derived from NCBI gene priors and contextualized Cell2Sentence embeddings, and a Structure-Aware Topological View extracted via a graph-guided encoder. Crucially, we introduce a cross-modal contrastive alignment mechanism to enforce consistency between biological semantics and transcriptomic features within a unified latent space. Extensive benchmarks demonstrate that scLLM-DSC significantly outperforms eleven state-of-the-art baselines in clustering accuracy.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes scLLM-DSC, a cross-modal deep structural clustering framework for scRNA-seq data. It constructs a Knowledge-Driven Semantic View from NCBI gene priors and Cell2Sentence embeddings, pairs it with a Structure-Aware Topological View from a graph-guided encoder, and uses cross-modal contrastive alignment to produce a unified latent space; the abstract asserts that this yields significant gains over eleven state-of-the-art baselines.
Significance. If the reported gains are substantiated by rigorous, reproducible experiments, the work would demonstrate a concrete route for injecting biological semantics into single-cell clustering, moving beyond purely statistical pattern mining. The cross-modal contrastive mechanism is a plausible way to address the generative-to-discriminative mismatch noted in the abstract.
major comments (2)
- [Abstract] Abstract: the central claim that 'Extensive benchmarks demonstrate that scLLM-DSC significantly outperforms eleven state-of-the-art baselines in clustering accuracy' is stated without any quantitative metrics, dataset names or sizes, ablation results, or description of how the contrastive alignment loss is formulated and validated. This absence makes the primary empirical contribution impossible to evaluate.
- [Abstract] Abstract: the description of the cross-modal contrastive alignment mechanism supplies no equations, temperature parameters, negative sampling strategy, or validation that the alignment actually improves downstream clustering rather than merely regularizing the latent space.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript. We address the major comments point by point below and will revise the abstract to incorporate more concrete details while preserving its summary nature.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that 'Extensive benchmarks demonstrate that scLLM-DSC significantly outperforms eleven state-of-the-art baselines in clustering accuracy' is stated without any quantitative metrics, dataset names or sizes, ablation results, or description of how the contrastive alignment loss is formulated and validated. This absence makes the primary empirical contribution impossible to evaluate.
Authors: We agree that the abstract would be more informative with specific quantitative support. In the revised version we will add key performance metrics (e.g., average ARI and NMI gains across the eleven baselines), the number and scale of the benchmark datasets, and a brief reference to the ablation studies and loss formulation that appear in Sections 3 and 4. These additions will make the empirical contribution clearer at the abstract level without duplicating the full experimental results. revision: yes
-
Referee: [Abstract] Abstract: the description of the cross-modal contrastive alignment mechanism supplies no equations, temperature parameters, negative sampling strategy, or validation that the alignment actually improves downstream clustering rather than merely regularizing the latent space.
Authors: Abstracts are not the appropriate venue for full equations, yet we accept that a more informative description is warranted. We will revise the abstract to include a concise statement of the contrastive alignment (temperature-scaled InfoNCE loss with in-batch negatives) and note that ablation experiments demonstrate its benefit to clustering accuracy. The complete formulation, parameter values, and validation appear in Section 3.3 of the manuscript. revision: yes
Circularity Check
No significant circularity identified
full rationale
The paper's abstract and description present a high-level framework for cross-modal contrastive alignment between LLM-derived semantic views (NCBI gene priors and Cell2Sentence embeddings) and transcriptomic features via a graph-guided encoder, with claimed outperformance on benchmarks. No equations, parameter-fitting procedures, derivation chains, or self-citations are visible in the provided text that reduce any prediction or result to its own inputs by construction. The method is described as establishing a unified latent space through alignment, but this is presented as a design choice rather than a self-referential or fitted tautology. The central claim rests on external benchmark comparisons, which are independent of internal circularity. This is the expected outcome for a methods paper without visible mathematical reductions.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Integrat- ing single-cell transcriptomic data across different condi- tions, technologies, and species.Nature biotechnology, 36(5):411–420,
[Butleret al., 2018 ] Andrew Butler, Paul Hoffman, Peter Smibert, Efthymia Papalexi, and Rahul Satija. Integrat- ing single-cell transcriptomic data across different condi- tions, technologies, and species.Nature biotechnology, 36(5):411–420,
2018
-
[2]
Genept: a simple but effective foundation model for genes and cells built from chatgpt.bioRxiv, pages 2023–10,
[Chen and Zou, 2024] Yiqun Chen and James Zou. Genept: a simple but effective foundation model for genes and cells built from chatgpt.bioRxiv, pages 2023–10,
2024
-
[3]
Deep soft k-means clustering with self- training for single-cell rna sequence data.NAR genomics and bioinformatics, 2(2):lqaa039,
[Chenet al., 2020 ] Liang Chen, Weinan Wang, Yuyao Zhai, and Minghua Deng. Deep soft k-means clustering with self- training for single-cell rna sequence data.NAR genomics and bioinformatics, 2(2):lqaa039,
2020
-
[4]
scgpt: toward building a foundation model for single-cell multi- omics using generative ai.Nature methods, 21(8):1470– 1480,
[Cuiet al., 2024 ] Haotian Cui, Chloe Wang, Hassaan Maan, Kuan Pang, Fengning Luo, Nan Duan, and Bo Wang. scgpt: toward building a foundation model for single-cell multi- omics using generative ai.Nature methods, 21(8):1470– 1480,
2024
-
[5]
[Dipet al., 2025 ] Sajib Acharjee Dip, Adrika Zafor, Bikash Kumar Paul, Uddip Acharjee Shuvo, Muhit Islam Emon, Xuan Wang, and Liqing Zhang. Llm4cell: A survey of large language and agentic models for single-cell biology.arXiv preprint arXiv:2510.07793,
-
[6]
Single- cell rna-seq denoising using a deep count autoencoder.Na- ture communications, 10(1):390,
[Eraslanet al., 2019 ] Gökcen Eraslan, Lukas M Simon, Maria Mircea, Nikola S Mueller, and Fabian J Theis. Single- cell rna-seq denoising using a deep count autoencoder.Na- ture communications, 10(1):390,
2019
-
[7]
scmae: a masked autoencoder for single-cell rna-seq clustering.Bioinformatics, 40(1):btae020,
[Fanget al., 2024 ] Zhaoyu Fang, Ruiqing Zheng, and Min Li. scmae: a masked autoencoder for single-cell rna-seq clustering.Bioinformatics, 40(1):btae020,
2024
-
[8]
Deep structural clustering for single-cell rna-seq data jointly through au- toencoder and graph neural network.Briefings in Bioinfor- matics, 23(2):bbac018,
[Ganet al., 2022 ] Yanglan Gan, Xingyu Huang, Guobing Zou, Shuigeng Zhou, and Jihong Guan. Deep structural clustering for single-cell rna-seq data jointly through au- toencoder and graph neural network.Briefings in Bioinfor- matics, 23(2):bbac018,
2022
-
[9]
Large-scale foundation model on single-cell transcriptomics.Nature methods, 21(8):1481–1491,
[Haoet al., 2024 ] Minsheng Hao, Jing Gong, Xin Zeng, Chiming Liu, Yucheng Guo, Xingyi Cheng, Taifeng Wang, Jianzhu Ma, Xuegong Zhang, and Le Song. Large-scale foundation model on single-cell transcriptomics.Nature methods, 21(8):1481–1491,
2024
-
[10]
Zero-shot evaluation re- veals limitations of single-cell foundation models.Genome Biology, 26(1):101,
[Kedzierskaet al., 2025 ] Kasia Z Kedzierska, Lorin Craw- ford, Ava P Amini, and Alex X Lu. Zero-shot evaluation re- veals limitations of single-cell foundation models.Genome Biology, 26(1):101,
2025
-
[11]
Challenges in unsupervised clustering of single-cell rna-seq data.Nature Reviews Ge- netics, 20(5):273–282,
[Kiselevet al., 2019 ] Vladimir Yu Kiselev, Tallulah S An- drews, and Martin Hemberg. Challenges in unsupervised clustering of single-cell rna-seq data.Nature Reviews Ge- netics, 20(5):273–282,
2019
-
[12]
Cell2sentence: teaching large language models the language of biology.BioRxiv, pages 2023–09,
[Levineet al., 2024 ] Daniel Levine, Syed Asad Rizvi, Sacha Lévy, Nazreen Pallikkavaliyaveetil, David Zhang, Xingyu Chen, Sina Ghadermarzi, Ruiming Wu, Zihe Zheng, Ivan Vrkic, et al. Cell2sentence: teaching large language models the language of biology.BioRxiv, pages 2023–09,
2024
-
[13]
screader: Prompting large language mod- els to interpret scrna-seq data
[Liet al., 2024 ] Cong Li, Qingqing Long, Yuanchun Zhou, and Meng Xiao. screader: Prompting large language mod- els to interpret scrna-seq data. In2024 IEEE International Conference on Data Mining Workshops (ICDMW), pages 665–672. IEEE,
2024
-
[14]
Sceval: An open-source platform for standardized evaluation and optimization of standard cell libraries in next-generation process nodes
[Liet al., 2025 ] Longfan Li, Wangzilu Lu, Yuxin Ji, Zhiwen Gu, Huajie Huang, Yuhang Zhang, Jian Zhao, and Yongfu Li. Sceval: An open-source platform for standardized evaluation and optimization of standard cell libraries in next-generation process nodes. In2025 International Con- ference on Electronics, Information, and Communication (ICEIC), pages 1–4. IEEE,
2025
-
[15]
Deep generative modeling for single-cell transcriptomics
[Lopezet al., 2018 ] Romain Lopez, Jeffrey Regier, Michael B Cole, Michael I Jordan, and Nir Yosef. Deep generative modeling for single-cell transcriptomics. Nature methods, 15(12):1053–1058,
2018
-
[16]
Visualizing data using t-SNE.Journal of machine learning research, 9(Nov):2579–2605,
[Maaten and Hinton, 2008] Laurens van der Maaten and Ge- offrey Hinton. Visualizing data using t-SNE.Journal of machine learning research, 9(Nov):2579–2605,
2008
-
[17]
Exponential scaling of single-cell rna-seq in the past decade.Nature protocols, 13(4):599–604,
[Svenssonet al., 2018 ] Valentine Svensson, Roser Vento- Tormo, and Sarah A Teichmann. Exponential scaling of single-cell rna-seq in the past decade.Nature protocols, 13(4):599–604,
2018
-
[18]
Transfer learning enables predictions in network biology.Nature, 618(7965):616– 624,
[Theodoriset al., 2023 ] Christina V Theodoris, Ling Xiao, Anant Chopra, Mark D Chaffin, Zeina R Al Sayed, Matthew C Hill, Helene Mantineo, Elizabeth M Brydon, Zexian Zeng, X Shirley Liu, et al. Transfer learning enables predictions in network biology.Nature, 618(7965):616– 624,
2023
-
[19]
Clustering single-cell rna-seq data with a model-based deep learning approach.Nature Machine Intelligence, 1(4):191– 198,
[Tianet al., 2019 ] Tian Tian, Ji Wan, Qi Song, and Zhi Wei. Clustering single-cell rna-seq data with a model-based deep learning approach.Nature Machine Intelligence, 1(4):191– 198,
2019
-
[20]
Model-based deep embedding for constrained clustering analysis of single cell rna-seq data
[Tianet al., 2021 ] Tian Tian, Jie Zhang, Xiang Lin, Zhi Wei, and Hakon Hakonarson. Model-based deep embedding for constrained clustering analysis of single cell rna-seq data. Nature communications, 12(1):1873,
2021
-
[21]
scname: neighborhood contrastive clustering with ancil- lary mask estimation for scrna-seq data.Bioinformatics, 38(6):1575–1583,
[Wanet al., 2022 ] Hui Wan, Liang Chen, and Minghua Deng. scname: neighborhood contrastive clustering with ancil- lary mask estimation for scrna-seq data.Bioinformatics, 38(6):1575–1583,
2022
-
[22]
scgnn is a novel graph neural network framework for single-cell rna-seq analyses.Nature communications, 12(1):1882,
[Wanget al., 2021 ] Juexin Wang, Anjun Ma, Yuzhou Chang, Jianting Gong, Yuexu Jiang, Ren Qi, Cankun Wang, Hongjun Fu, Qin Ma, and Dong Xu. scgnn is a novel graph neural network framework for single-cell rna-seq analyses.Nature communications, 12(1):1882,
2021
-
[23]
Scanpy: large-scale single-cell gene expres- sion data analysis.Genome biology, 19:1–5,
[Wolfet al., 2018 ] F Alexander Wolf, Philipp Angerer, and Fabian J Theis. Scanpy: large-scale single-cell gene expres- sion data analysis.Genome biology, 19:1–5,
2018
-
[24]
sccdcg: efficient deep structural clustering for single-cell rna-seq via deep cut-informed graph embedding
[Xuet al., 2024 ] Ping Xu, Zhiyuan Ning, Meng Xiao, Guihai Feng, Xin Li, Yuanchun Zhou, and Pengfei Wang. sccdcg: efficient deep structural clustering for single-cell rna-seq via deep cut-informed graph embedding. InInternational Conference on Database Systems for Advanced Applica- tions, pages 172–187. Springer,
2024
-
[25]
scsiameseclu: A siamese clustering framework for interpreting single-cell rna sequencing data
[Xuet al., 2025a ] Ping Xu, Zhiyuan Ning, Pengjiang Li, Wenhao Liu, Pengyang Wang, Jiaxu Cui, Yuanchun Zhou, and Pengfei Wang. scsiameseclu: A siamese clustering framework for interpreting single-cell rna sequencing data. arXiv preprint arXiv:2505.12626,
-
[26]
scclubench: Comprehensive benchmark- ing of clustering algorithms for single-cell rna sequencing
[Xuet al., 2025c ] Ping Xu, Zaitian Wang, Zhirui Wang, Pengjiang Li, Jiajia Wang, Ran Zhang, Pengfei Wang, and Yuanchun Zhou. scclubench: Comprehensive benchmark- ing of clustering algorithms for single-cell rna sequencing. arXiv preprint arXiv:2512.02471,
-
[27]
[Xuet al., 2025d ] Ping Xu, Zaitian Wang, Zhirui Wang, Pengjiang Li, Ran Zhang, Gaoyang Li, Hanyu Xie, Jia- jia Wang, Yuanchun Zhou, and Pengfei Wang. scunified: An ai-ready standardized resource for single-cell rna se- quencing analysis.arXiv preprint arXiv:2509.25884,
-
[28]
scbert as a large-scale pretrained deep language model for cell type annotation of single-cell rna-seq data
[Yanget al., 2022 ] Fan Yang, Wenchuan Wang, Fang Wang, Yuan Fang, Duyu Tang, Junzhou Huang, Hui Lu, and Jian- hua Yao. scbert as a large-scale pretrained deep language model for cell type annotation of single-cell rna-seq data. Nature Machine Intelligence, 4(10):852–866,
2022
-
[29]
Genecom- pass: deciphering universal gene regulatory mechanisms with a knowledge-informed cross-species foundation model
[Yanget al., 2024 ] Xiaodong Yang, Guole Liu, Guihai Feng, Dechao Bu, Pengfei Wang, Jie Jiang, Shubai Chen, Qin- meng Yang, Hefan Miao, Yiyang Zhang, et al. Genecom- pass: deciphering universal gene regulatory mechanisms with a knowledge-informed cross-species foundation model. Cell Research, pages 1–16,
2024
-
[30]
[Yuanet al., 2025 ] Zhen Yuan, Shaoqing Jiao, Yihang Xiao, and Jiajie Peng. scmamba: A scalable foundation model for single-cell multi-omics integration beyond highly variable feature selection.arXiv preprint arXiv:2506.20697,
-
[31]
A survey on foundation language models for single-cell biology
[Zhanget al., 2025 ] Fan Zhang, Hao Chen, Zhihong Zhu, Zi- heng Zhang, Zhenxi Lin, Ziyue Qiao, Yefeng Zheng, and Xian Wu. A survey on foundation language models for single-cell biology. InProceedings of the 63rd Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers), pages 528–549, 2025
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.