Machine learning-based modeling to predict inhibitors for targets of Alzheimer's Disease
Pith reviewed 2026-06-25 21:36 UTC · model grok-4.3
The pith
Machine learning models predict inhibitors for Alzheimer's targets BACE-1, AChE, and GSK-3 beta with AUC-ROC scores above 0.9.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
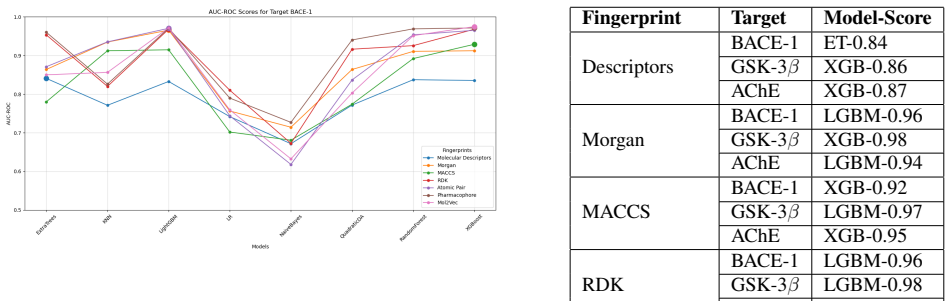
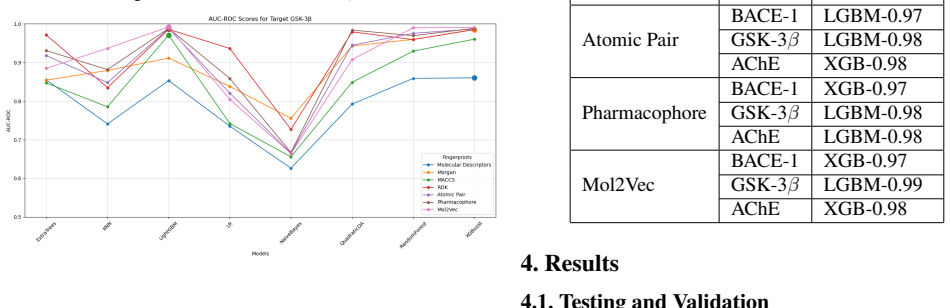
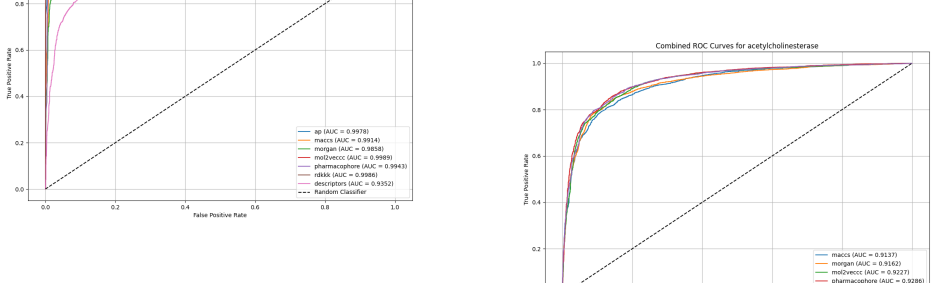
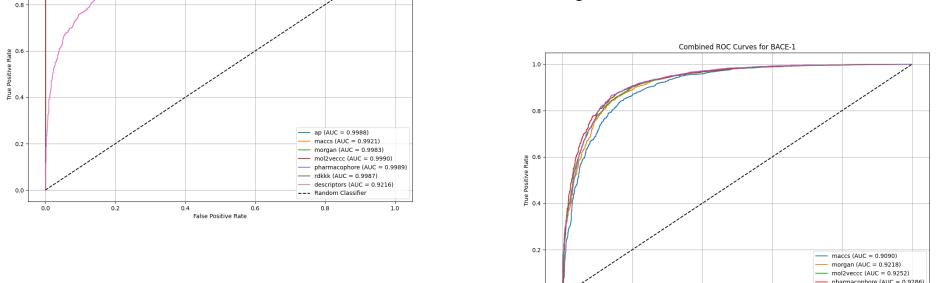
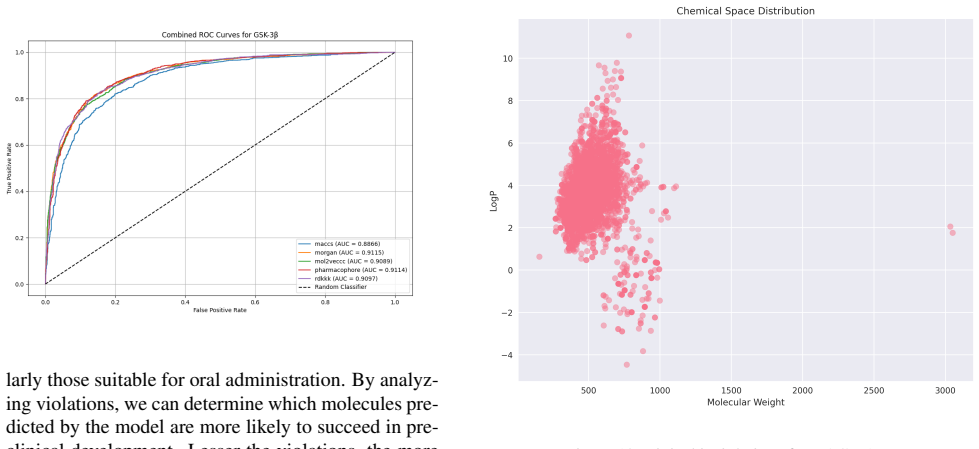
Utilizing machine learning, predictive models for inhibitor screening were developed, achieving AUC-ROC scores above 0.9 for all targets. BACE-1 models showed high accuracy (86.63%) but limited chemical diversity. AChE models exhibited greater chemical diversity and similar performance (AUC-ROC: 92.86%, Accuracy: 85.20%), while GSK-3 beta models achieved an AUC-ROC of 91.14% with the highest proportion of viable drug candidates. These findings highlight the potential of ML in Alzheimer's drug discovery, with AChE and GSK-3 beta emerging as promising targets.
What carries the argument
Machine learning classification models trained on molecular features of chemical compounds to distinguish inhibitors from non-inhibitors for Alzheimer's targets.
If this is right
- AChE and GSK-3 beta emerge as promising targets for further drug development based on model diversity and candidate yield.
- ML-based virtual screening can be applied to identify inhibitor candidates for these Alzheimer's targets.
- GSK-3 beta models produced the highest proportion of viable drug candidates among the three.
- The approach demonstrates the utility of ML for inhibitor prediction in neurodegenerative disease targets.
Where Pith is reading between the lines
- If the models generalize beyond the training set, they could prioritize compounds for experimental testing and reduce the number of compounds that need physical screening.
- The reported chemical diversity differences suggest that target-specific training data quality directly affects how broadly the models can be applied.
- Extending the models to include additional Alzheimer's-related targets or combining predictions across targets might improve overall hit rates.
Load-bearing premise
High performance measured on internal test sets from the training data will translate to accurate predictions for new compounds outside that chemical space.
What would settle it
Biochemical or cell-based assays that measure whether the top predicted viable candidates actually inhibit the enzymatic activity of BACE-1, AChE, or GSK-3 beta at expected concentrations.
Figures





read the original abstract
Alzheimer's Disease is a chronic neurodegenerative disorder projected to affect 115 million people by 2050, driven by mechanisms like the cholinergic and amyloid hypotheses and insulin signaling disruptions involving key targets such as BACE-1, AChE, and GSK-3 beta. Utilizing machine learning (ML), we developed predictive models for inhibitor screening, achieving AUC-ROC scores above 0.9 for all targets. BACE-1 models showed high accuracy (86.63%) but limited chemical diversity. AChE models exhibited greater chemical diversity and similar performance (AUC-ROC: 92.86%, Accuracy: 85.20%), while GSK-3 beta models achieved an AUC-ROC of 91.14% with the highest proportion of viable drug candidates. These findings highlight the potential of ML in Alzheimer's drug discovery, with AChE and GSK-3 beta emerging as promising targets.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript applies machine learning to develop predictive models for identifying inhibitors of three Alzheimer's disease targets (BACE-1, AChE, and GSK-3β). It reports AUC-ROC scores above 0.9 for all targets along with accuracies of 86.63% (BACE-1) and 85.20% (AChE), notes differences in chemical diversity across targets, and concludes that the models highlight the potential of ML for AD drug discovery with AChE and GSK-3β as promising targets.
Significance. If the performance claims hold under proper validation, the work would demonstrate standard supervised learning applied to established AD targets, but the absence of any mention of reproducible code, parameter-free derivations, or prospective validation means no special credit applies on those dimensions. The central empirical claims cannot be assessed for impact without the missing methodological details.
major comments (3)
- [Abstract] Abstract: the central claim that AUC-ROC scores above 0.9 were achieved supplies no information on dataset size, feature engineering, cross-validation strategy, or chemical diversity handling; without these the performance numbers cannot be evaluated and the screening utility claim is unsupported.
- [Abstract] Abstract: no description is given of the train/test split methodology (random, scaffold, temporal, etc.), whether test compounds lie inside or outside the training chemical distribution, or any external/prospective validation; this directly undermines the use-case claim that the models will identify viable inhibitors for novel compounds.
- [Abstract] Abstract: the statement that GSK-3β models yielded 'the highest proportion of viable drug candidates' is presented without any definition of 'viable,' experimental binding data, or comparison to known actives, rendering the ranking across targets unevaluable.
minor comments (1)
- [Abstract] The abstract mentions 'limited chemical diversity' for BACE-1 and 'greater chemical diversity' for AChE/GSK-3β but provides no quantitative measure (e.g., Tanimoto similarity distributions or scaffold counts).
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on the abstract. We agree that additional methodological context is needed for proper evaluation of the reported performance metrics and will revise the abstract accordingly while preserving the manuscript's core findings. Details on datasets, validation, and definitions are present in the full text but will be summarized in the abstract for completeness.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that AUC-ROC scores above 0.9 were achieved supplies no information on dataset size, feature engineering, cross-validation strategy, or chemical diversity handling; without these the performance numbers cannot be evaluated and the screening utility claim is unsupported.
Authors: We agree the abstract should be more informative. The full manuscript reports dataset sizes (number of active/inactive compounds per target), ECFP fingerprints for feature engineering, 5-fold cross-validation, and chemical diversity assessed via Tanimoto similarity and scaffold analysis. We will revise the abstract to include brief statements on these elements so the AUC-ROC and accuracy figures can be evaluated in context. revision: yes
-
Referee: [Abstract] Abstract: no description is given of the train/test split methodology (random, scaffold, temporal, etc.), whether test compounds lie inside or outside the training chemical distribution, or any external/prospective validation; this directly undermines the use-case claim that the models will identify viable inhibitors for novel compounds.
Authors: The manuscript uses a random 80/20 train/test split with internal 5-fold cross-validation; test compounds are within the same chemical distribution as the training set (confirmed via similarity analysis). No external or prospective validation is described because none was performed. We will add this clarification to the abstract and note that the screening utility claim is based on internal validation performance. revision: yes
-
Referee: [Abstract] Abstract: the statement that GSK-3β models yielded 'the highest proportion of viable drug candidates' is presented without any definition of 'viable,' experimental binding data, or comparison to known actives, rendering the ranking across targets unevaluable.
Authors: We acknowledge that 'viable' requires definition. In the manuscript, viable drug candidates refer to compounds predicted as inhibitors with probability >0.8 that also satisfy Lipinski's rule-of-five criteria. This is purely in silico and not supported by experimental binding data or direct comparison to known actives beyond the training set. We will revise the abstract to explicitly define the term and qualify the ranking as based on predicted properties only. revision: yes
Circularity Check
No circularity: empirical ML evaluation with no derivations or self-referential reductions
full rationale
The paper reports training ML classifiers on compound datasets for three Alzheimer's targets and evaluating them via standard metrics (AUC-ROC, accuracy) on held-out test sets. No equations, ansatzes, uniqueness theorems, or derivation chains appear in the provided text. Performance numbers are obtained by fitting models to training data and scoring on separate test compounds; they are not redefined or forced by the inputs themselves. No self-citations are invoked to justify core premises. The work is therefore self-contained as an empirical modeling study, and the reported results do not reduce to their own inputs by construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Hardeep Sandhu and Rajaram Naresh Kumar and Prabha Garg , title =. Molecular Diversity , year =. doi:10.1007/s11030-021-10223-5 , issn =
-
[2]
G. Dhamodharan and C. Gopi Mohan , title =. Molecular Diversity , year =. doi:10.1007/s11030-021-10282-8 , issn =
-
[3]
Drug treatments in Alzheimer’s disease , journal =
Robert Briggs and Sean P Kennelly and Desmond O’Neill , keywords =. Drug treatments in Alzheimer’s disease , journal =. 2016 , issn =. doi:https://doi.org/10.7861/clinmedicine.16-3-247 , url =
-
[4]
Heca Journal of Applied Sciences , volume=
QSAR Classification of Beta-Secretase 1 Inhibitor Activity in Alzheimer's Disease Using Ensemble Machine Learning Algorithms , author=. Heca Journal of Applied Sciences , volume=
-
[5]
International Journal of Molecular Sciences , VOLUME =
Galati, Salvatore and Di Stefano, Miriana and Bertini, Simone and Granchi, Carlotta and Giordano, Antonio and Gado, Francesca and Macchia, Marco and Tuccinardi, Tiziano and Poli, Giulio , TITLE =. International Journal of Molecular Sciences , VOLUME =. 2023 , NUMBER =
2023
-
[6]
Scientific reports , volume=
QSAR classification models for predicting the activity of inhibitors of beta-secretase (BACE1) associated with Alzheimer’s disease , author=. Scientific reports , volume=. 2019 , publisher=
2019
-
[7]
Zdrazil, Barbara and Felix, Eloy and Hunter, Fiona and Manners, Emma J and Blackshaw, James and Corbett, Sybilla and de Veij, Marleen and Ioannidis, Harris and Lopez, David Mendez and Mosquera, Juan F and Magarinos, Maria Paula and Bosc, Nicolas and Arcila, Ricardo and Kizilören, Tevfik and Gaulton, Anna and Bento, A Patrícia and Adasme, Melissa F and Mon...
-
[8]
Liu, T. and Lin, Y. and Wen, X. and Jorissen, R. N. and Gilson, M. K. , title =. Nucleic Acids Research , year =. doi:10.1093/nar/gkl999 , url =
-
[9]
and Tian, Y.-S
Moriwaki, H. and Tian, Y.-S. and Kawashita, N. and Takagi, T. , title =. Journal of Cheminformatics , year =
-
[10]
2020 , note =
Greg Landrum and Paolo Tosco and Brian Kelley and sriniker and gedeck and Nadine Schneider and Riccardo Vianello and Ric and Andrew Dalke and Brian Cole and Alexander Savelyev and Matt Swain and Samo Turk and Dan N and Alain Vaucher and Eisuke Kawashima and Maciej Wójcikowski and Daniel Probst and guillaume godin and David Cosgrove and Axel Pahl and JP an...
2020
-
[11]
Journal of Chemical Information and Modeling , year=
Extended-Connectivity Fingerprints , author=. Journal of Chemical Information and Modeling , year=
-
[12]
Journal of Chemical Information and Computer Sciences , year=
Reoptimization of MDL Keys for Use in Drug Discovery , author=. Journal of Chemical Information and Computer Sciences , year=
-
[13]
Journal of Chemical Information and Computer Sciences , year=
Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications , author=. Journal of Chemical Information and Computer Sciences , year=
-
[14]
Journal of Medicinal Chemistry , year=
Pharmacophore Fingerprints: A New Tool for Searching Molecular Databases , author=. Journal of Medicinal Chemistry , year=
-
[15]
Comparison with other Descriptors , author=
Topological Torsion: A New Molecular Descriptor for SAR Applications. Comparison with other Descriptors , author=. Journal of Chemical Information and Computer Sciences , year=
-
[16]
Journal of Chemical Information and Modeling , year=
Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition , author=. Journal of Chemical Information and Modeling , year=
-
[17]
Advances in Intelligent Computing , volume=
Borderline-SMOTE: A New Over-Sampling Method in Imbalanced Data Sets Learning , author=. Advances in Intelligent Computing , volume=. 2005 , publisher=
2005
-
[18]
Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining , pages=
Optuna: A Next-generation Hyperparameter Optimization Framework , author=. Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining , pages=. 2019 , publisher=
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.