MALOQ: Massively Accelerated Learning of Operators for Quantum Transport
Pith reviewed 2026-06-30 09:59 UTC · model grok-4.3
The pith
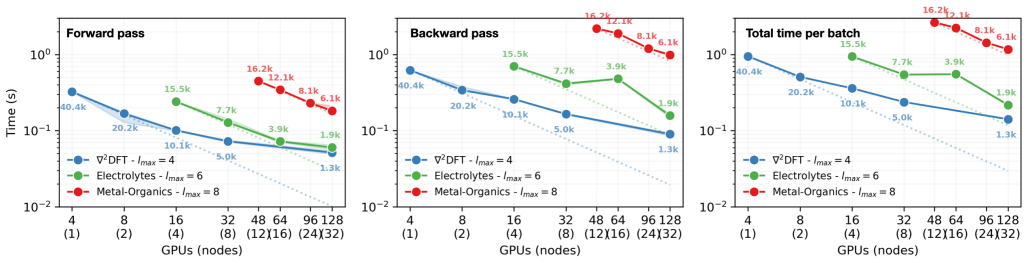
MALOQ trains equivariant models on the largest Hamiltonian datasets to predict quantum operators for systems up to 100,000 atoms while cutting time-per-epoch by over 30 percent.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
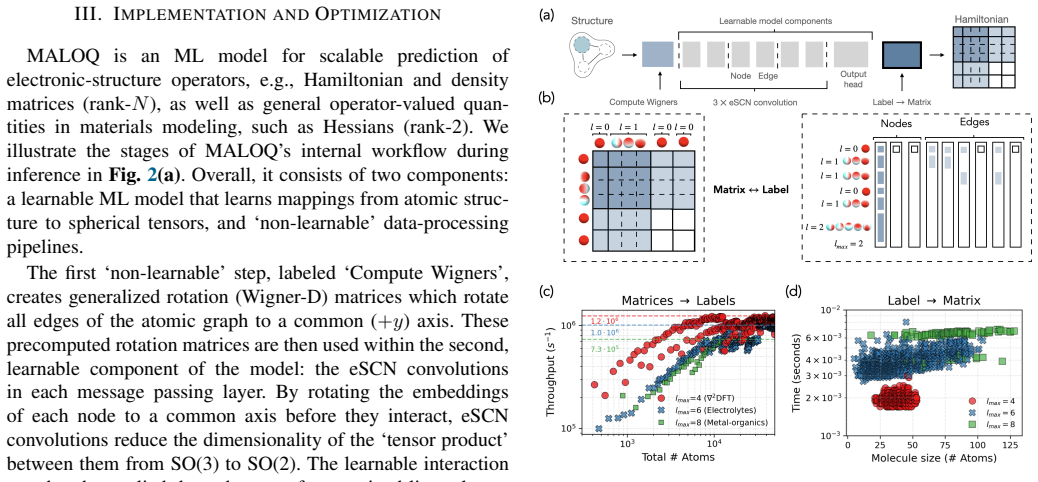
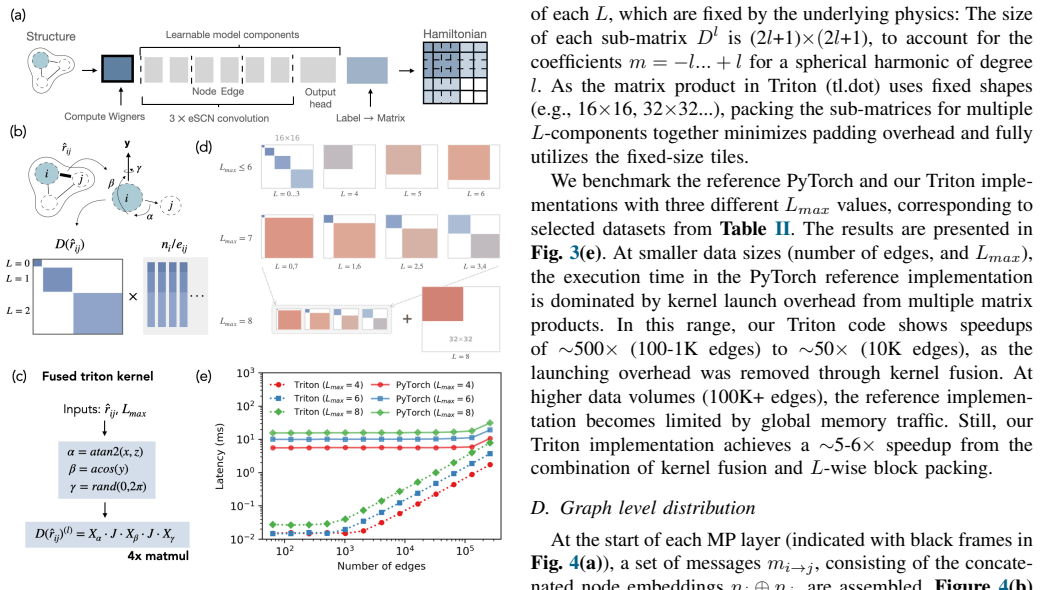
MALOQ is an application that trains SO(2)-equivariant models on large molecular Hamiltonian datasets to predict electronic-structure matrices for systems of a few to 100k atoms, using custom data-processing kernels for high-rank matrices and a scalable edge-wise distribution of atomic graphs, thereby reducing time-per-epoch by more than 30 percent relative to molecule-wise distribution and enabling inference on material graphs of arbitrary size.
What carries the argument
SO(2)-equivariant backbone architecture together with custom data-processing kernels for high-rank Hamiltonian matrices and an edge-wise distribution scheme for atomic graphs.
If this is right
- Training on the largest available molecular Hamiltonian datasets becomes feasible with more than 30 percent lower time per epoch.
- Inference runs on material graphs whose total atom count is no longer constrained by the size of the training examples.
- Electronic-structure calculations extend to system sizes previously unreachable by direct DFT methods.
- Scalable training and inference are demonstrated for 3,000-12,000 atom systems on up to 256 GPUs.
Where Pith is reading between the lines
- The same distribution and kernel approach could be reused for other high-rank quantum operators such as density matrices or current operators.
- Once trained, the model could serve as a fast surrogate inside device-scale transport simulations that mix atomistic and continuum regions.
- The arbitrary-size inference property removes the need to retrain when moving from molecular test sets to periodic or nanostructured materials.
Load-bearing premise
The SO(2)-equivariant model with the custom kernels learns accurate high-rank Hamiltonian predictions across many atomic elements and system sizes up to 100k atoms without substantial accuracy loss.
What would settle it
A direct accuracy comparison on a held-out set of 50,000-atom structures containing underrepresented elements that shows prediction errors growing beyond the tolerance reported for smaller training systems.
Figures





read the original abstract
Machine-learned (ML) operator models can be trained to predict density functional theory (DFT) Hamiltonian/density matrices at significantly reduced computational cost, thus extending electronic-structure calculations to previously unfeasible scales. Here, we introduce MALOQ (Massively Accelerated Learning of Operators for Quantum Transport), an application built to train on and predict electronic-structure matrices for systems made of few to 100k atoms, described by large basis sets, and covering a wide range of atomic elements. Based on a state-of-the-art, SO(2)-equivariant backbone architecture, MALOQ provides (i) custom data-processing kernels to handle high-rank Hamiltonian matrix data and (ii) a scalable edge-wise distribution of atomic graph(s). Trained on the largest molecular Hamiltonian datasets available today, it reduces time-per-epoch by over 30% compared to a molecule-wise-distributed framework, and enables inference on material graphs of arbitrary size. We demonstrate scalable training and inference for 3,000-12,000 atoms on the Alps supercomputer, up to 192 GPUs and 256 GPUs, respectively.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces MALOQ, a framework built on an SO(2)-equivariant backbone with custom data-processing kernels and edge-wise graph distribution for training ML models to predict DFT Hamiltonian/density matrices. It claims >30% reduction in time-per-epoch versus molecule-wise distribution, scalable training/inference on 3k–12k atom systems (up to 192/256 GPUs on Alps), and the ability to handle arbitrary-size material graphs up to 100k atoms across diverse elements and large basis sets.
Significance. If accuracy on large systems is validated, the work could enable electronic-structure calculations at previously inaccessible scales for quantum transport. The reported engineering contributions (custom kernels, edge-wise distribution) address real bottlenecks in distributed training on high-rank matrix data, and the scaling demonstrations on a production supercomputer are concrete.
major comments (2)
- [Abstract] Abstract: the central utility claim—that the model learns accurate high-rank Hamiltonian predictions for systems up to 100k atoms without substantial accuracy loss—is unsupported; no MAE on matrix elements, eigenvalue errors, transport quantities, or DFT ground-truth comparisons are reported for any system size, including the 3k–12k atom demonstrations.
- [Abstract] Abstract and scaling sections: the assertion of inference on material graphs of arbitrary size rests on extrapolation from 3k–12k atom results; no evidence (accuracy metrics, ablation on graph size, or tests on non-molecular periodic systems) is provided to substantiate that accuracy holds at 100k atoms or for arbitrary-size inference.
minor comments (2)
- The manuscript should include at least one table or figure reporting quantitative accuracy (e.g., MAE per matrix element or band-structure error) against DFT reference for the largest demonstrated systems.
- Clarify the precise definition of 'time-per-epoch' reduction (wall-clock, per-GPU, or communication overhead) and the baseline molecule-wise framework implementation details.
Simulated Author's Rebuttal
Thank you for the referee's constructive feedback on our manuscript. We appreciate the identification of areas where the abstract and scaling claims require additional support or clarification. We address each major comment below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central utility claim—that the model learns accurate high-rank Hamiltonian predictions for systems up to 100k atoms without substantial accuracy loss—is unsupported; no MAE on matrix elements, eigenvalue errors, transport quantities, or DFT ground-truth comparisons are reported for any system size, including the 3k–12k atom demonstrations.
Authors: We agree that the abstract overstates the validated accuracy for large systems. The current work emphasizes the engineering contributions for scalable training and inference on high-rank matrix data, with accuracy inherited from the SO(2)-equivariant backbone trained on available datasets. To address this, the revised manuscript will include MAE on matrix elements, eigenvalue errors, and transport quantity comparisons for the 3k–12k atom demonstrations, along with explicit DFT ground-truth references where available. The 100k-atom claim will be qualified as prospective. revision: yes
-
Referee: [Abstract] Abstract and scaling sections: the assertion of inference on material graphs of arbitrary size rests on extrapolation from 3k–12k atom results; no evidence (accuracy metrics, ablation on graph size, or tests on non-molecular periodic systems) is provided to substantiate that accuracy holds at 100k atoms or for arbitrary-size inference.
Authors: We acknowledge that the arbitrary-size inference claim relies on the design of the edge-wise distribution, which removes molecule-wise batching constraints and permits processing of graphs of any size in principle. The 3k–12k atom results demonstrate linear scaling behavior up to the tested regime. In the revision, we will update the abstract and scaling sections to state that accuracy at 100k atoms and on non-molecular periodic systems is extrapolated rather than directly validated, and we will add a dedicated limitations paragraph on this point. revision: yes
Circularity Check
No circularity detected; claims rest on implementation and empirical scaling demos
full rationale
The paper describes an ML application (MALOQ) using an SO(2)-equivariant GNN backbone plus custom kernels for matrix data and edge-wise graph distribution. Performance claims (30% epoch time reduction, arbitrary-size inference) are presented as outcomes of these engineering choices, with concrete scaling results shown only for 3k-12k atom systems. No derivations, fitted parameters renamed as predictions, self-definitional loops, or load-bearing self-citations appear in the provided text. The 100k-atom accuracy claim is an unverified extrapolation but does not reduce any result to its inputs by construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Foundation models for atomistic simulation of chemistry and materials,
E. C.-Y . Yuan, Y . Liu, J. Chen, P. Zhong, S. Raja, T. Kreiman, S. Vargas, W. Xu, M. Head-Gordon, C. Yang, S. M. Blau, B. Cheng, A. Krishnapriyan, and T. Head-Gordon, “Foundation models for atomistic simulation of chemistry and materials,”Nature Reviews Chemistry, vol. 10, no. 3, p. 212–230, Feb. 2026. [Online]. Available: http://dx.doi.org/10.1038/s4157...
-
[2]
A foundation model for atomistic materials chemistry,
I. Batatia, P. Benner, Y . Chiang, A. M. Elena, D. P. Kov ´acs, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, F. Berger, N. Bernstein, A. Bhowmik, F. Bigi, S. M. Blau, V . C ˘arare, M. Ceriotti, S. Chong, J. P. Darby, S. De, F. Della Pia, V . L. Deringer, R. Elijo ˇsius, Z. El-Machachi, E. Fako, F. Falcioni, A. C. Ferrari, J. L. A. Ga...
-
[3]
UMA: A family of universal models for atoms,
B. M. Wood, M. Dzamba, X. Fu, M. Gao, M. Shuaibi, L. Barroso- Luque, K. Abdelmaqsoud, V . Gharakhanyan, J. R. Kitchin, D. S. Levine, K. Michel, A. Sriram, T. Cohen, A. Das, S. J. Sahoo, A. Rizvi, Z. W. Ulissi, and C. L. Zitnick, “UMA: A family of universal models for atoms,” inThe Thirty-ninth Annual Conference on Neural Information Processing Systems, 20...
2025
-
[4]
Pet-mad as a lightweight universal interatomic potential for advanced materials modeling,
A. Mazitov, F. Bigi, M. Kellner, P. Pegolo, D. Tisi, G. Fraux, S. Pozdnyakov, P. Loche, and M. Ceriotti, “Pet-mad as a lightweight universal interatomic potential for advanced materials modeling,” Nature Communications, vol. 16, no. 1, Nov. 2025. [Online]. Available: http://dx.doi.org/10.1038/s41467-025-65662-7 1, 3
-
[5]
General framework for E(3)-equivariant neural network representation of density functional theory hamiltonian,
X. Gong, H. Li, N. Zou, R. Xu, W. Duan, and Y . Xu, “General framework for E(3)-equivariant neural network representation of density functional theory hamiltonian,”Nature Communications, vol. 14, no. 1, May 2023. [Online]. Available: http://dx.doi.org/10. 1038/s41467-023-38468-8 1, 3, 10
2023
-
[6]
Learning from the electronic structure of molecules across the periodic table,
M. Kaniselvan, B. K. Miller, M. Gao, J. Nam, and D. S. Levine, “Learning from the electronic structure of molecules across the periodic table,” inThe F ourteenth International Conference on Learning Representations, 2026. [Online]. Available: https: //openreview.net/forum?id=PS1YS8Wv4t 1, 2, 3, 4, 5, 10
2026
-
[8]
Enhancing the scalability and applicability of kohn-sham hamiltonians for molecular systems,
Y . Li, Z. Xia, L. Huang, X. Wei, S. Harshe, H. Yang, E. Luo, Z. Wang, J. Zhang, C. Liu, B. Shao, and M. Gerstein, “Enhancing the scalability and applicability of kohn-sham hamiltonians for molecular systems,” inThe Thirteenth International Conference on Learning Representations, 2025. [Online]. Available: https: //openreview.net/forum?id=twEvvkQqPS 1, 3, 4, 10
2025
-
[9]
Unified deep learning framework for many-body quantum chemistry via green’s functions,
C. Venturella, J. Li, C. Hillenbrand, X. Leyva Peralta, J. Liu, and T. Zhu, “Unified deep learning framework for many-body quantum chemistry via green’s functions,”Nature Computational Science, Jun. 2025. [Online]. Available: http://dx.doi.org/10.1038/s43588-025-00810-z 1
-
[11]
Density-functional method for nonequilibrium electron transport,
M. Brandbyge, J.-L. Mozos, P. Ordej ´on, J. Taylor, and K. Stokbro, “Density-functional method for nonequilibrium electron transport,” Physical Review B, vol. 65, no. 16, Mar. 2002. [Online]. Available: http://dx.doi.org/10.1103/PhysRevB.65.165401 1
-
[12]
N. Vetsch, A. Maeder, V . Maillou, A. Winka, J. Cao, G. Kwasniewski, L. Deuschle, T. Hoefler, A. N. Ziogas, and M. Luisier, “Ab-initio quantum transport with the gw approximation, 42, 240 atoms, and sustained exascale performance,” inProceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, ser. SC ’25. ...
-
[13]
Silicon ribbonfet cmos at 6nm gate length,
A. Agrawal, W. Chakraborty, W. Li, H. Ryu, B. Markman, S. H. Hoon, R. K. Paul, C. Y . Huang, S. M. Choi, K. Rho, A. Shu, R. Iglesias, P. Wallace, S. Ghosh, K. L. Cheong, J. L. Hockel, R. Thorman, L. Baumgartel, L. Shoer, V . Mishra, S. Berrada, A. Ashita, C. Weber, B. Obradovic, A. A. Oni, Z. Brooks, N. Franco, J. Kavalieros, and G. Dewey, “Silicon ribbon...
-
[14]
Approaching coupled-cluster accuracy for molecular electronic structures with multi-task learning,
H. Tang, B. Xiao, W. He, P. Subasic, A. R. Harutyunyan, Y . Wang, F. Liu, H. Xu, and J. Li, “Approaching coupled-cluster accuracy for molecular electronic structures with multi-task learning,”Nature Computational Science, vol. 5, no. 2, p. 144–154, Dec. 2024. [Online]. Available: http://dx.doi.org/10.1038/s43588-024-00747-9 1
-
[15]
Learning the electronic hamiltonian of large atomic structures,
C. H. Xia, M. Kaniselvan, A. N. Ziogas, M. Mladenovi ´c, R. Mahjoub, A. Maeder, and M. Luisier, “Learning the electronic hamiltonian of large atomic structures,” inF orty-second International Conference on Machine Learning, 2025. [Online]. Available: https://openreview.net/ forum?id=WGejWCgrpD 1, 2, 4, 10
2025
-
[17]
Orbitall: A unified quantum mechanical representation deep learning framework for all molecular systems,
B. S. Kang, V . C. Bhethanabotla, A. Tavakoli, M. D. Hanisch, W. A. Goddard, and A. Anandkumar, “Orbitall: A unified quantum mechanical representation deep learning framework for all molecular systems,”
-
[19]
Exploring the design space of machine learning models for quantum chemistry with a fully differentiable framework,
D. Suman, J. Nigam, S. Saade, P. Pegolo, H. T ¨urk, X. Zhang, G. K.-L. Chan, and M. Ceriotti, “Exploring the design space of machine learning models for quantum chemistry with a fully differentiable framework,” Journal of Chemical Theory and Computation, vol. 21, no. 13, p. 6505–6516, Jun. 2025. [Online]. Available: http://dx.doi.org/10.1021/ acs.jctc.5c00522 1
2025
-
[21]
Reducing SO(3) convolutions to SO(2) for efficient equivariant GNNs,
S. Passaro and C. L. Zitnick, “Reducing SO(3) convolutions to SO(2) for efficient equivariant GNNs,” inProceedings of the 40th International Conference on Machine Learning, ser. Proceedings of Machine Learning Research, A. Krause, E. Brunskill, K. Cho, B. Engelhardt, S. Sabato, and J. Scarlett, Eds., vol. 202. PMLR, 23–29 Jul 2023, pp. 27 420–27 438. [Onl...
2023
-
[22]
Learning smooth and expressive interatomic potentials for physical property prediction,
X. Fu, B. M. Wood, L. Barroso-Luque, D. S. Levine, M. Gao, M. Dzamba, and C. L. Zitnick, “Learning smooth and expressive interatomic potentials for physical property prediction,” inProceedings of the 42nd International Conference on Machine Learning, ser. Proceedings of Machine Learning Research, A. Singh, M. Fazel, D. Hsu, S. Lacoste-Julien, F. Berkenkam...
2025
-
[23]
Self-consistent equations including exchange and correlation effects,
W. Kohn and L. J. Sham, “Self-consistent equations including exchange and correlation effects,”Physical Review, vol. 140, no. 4A, p. A1133–A1138, Nov. 1965. [Online]. Available: http: //dx.doi.org/10.1103/PhysRev.140.A1133 3
-
[24]
T. D. K ¨uhne, M. Iannuzzi, M. Del Ben, V . V . Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Sch ¨utt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V . Weber, U. Bor ˇstnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. M ¨uller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Heh...
-
[25]
F. Neese, “The orca program system,”WIREs Computational Molecular Science, vol. 2, no. 1, p. 73–78, Jun. 2011. [Online]. Available: http://dx.doi.org/10.1002/wcms.81 3
-
[26]
W. Jia, H. Wang, M. Chen, D. Lu, L. Lin, R. Car, W. E, and L. Zhang, “Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning,” inProceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, ser. SC ’20. IEEE Press, 2020. [Online]. Available: https://dl.acm...
-
[27]
B. Kozinsky, A. Musaelian, A. Johansson, and S. Batzner, “Scaling the leading accuracy of deep equivariant models to biomolecular simulations of realistic size,” inProceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, ser. SC ’23. ACM, Nov. 2023, p. 1–12. [Online]. Available: http://dx.doi.org/10.114...
-
[28]
Y . Park, J. Kim, S. Hwang, and S. Han, “Scalable parallel algorithm for graph neural network interatomic potentials in molecular dynamics simulations,”Journal of Chemical Theory and Computation, vol. 20, no. 11, p. 4857–4868, May 2024. [Online]. Available: http://dx.doi.org/10.1021/acs.jctc.4c00190 3, 4
-
[29]
Efficient and equivariant graph networks for predicting quantum Hamiltonian,
H. Yu, Z. Xu, X. Qian, X. Qian, and S. Ji, “Efficient and equivariant graph networks for predicting quantum Hamiltonian,” inProceedings of the 40th International Conference on Machine Learning, ser. Proceedings of Machine Learning Research, A. Krause, E. Brunskill, K. Cho, B. Engelhardt, S. Sabato, and J. Scarlett, Eds., vol. 202. PMLR, 23–29 Jul 2023, pp...
2023
-
[31]
URL https://doi.org/10.1038/s41467-022-29939-5
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, “E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials,” Nature Communications, vol. 13, no. 1, May 2022. [Online]. Available: http://dx.doi.org/10.1038/s41467-022-29939-5 3, 4
-
[32]
Does equivariance matter at scale?
J. Brehmer, S. Behrends, P. D. Haan, and T. Cohen, “Does equivariance matter at scale?”Transactions on Machine Learning Research, 2025. [Online]. Available: https://openreview.net/forum?id=wilNute8Tn 3
2025
-
[33]
K. T. Sch ¨utt, M. Gastegger, A. Tkatchenko, K.-R. M ¨uller, and R. J. Maurer, “Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions,”Nature Communications, vol. 10, no. 1, Nov. 2019. [Online]. Available: http://dx.doi.org/10.1038/s41467-019-12875-2 4
-
[34]
Qh9: A quantum hamiltonian prediction benchmark for qm9 molecules,
H. Yu, M. Liu, Y . Luo, A. Strasser, X. Qian, X. Qian, and S. Ji, “Qh9: A quantum hamiltonian prediction benchmark for qm9 molecules,” inAdvances in Neural Information Processing Systems, A. Oh, T. Naumann, A. Globerson, K. Saenko, M. Hardt, and S. Levine, Eds., vol. 36. Curran Associates, Inc., 2023, pp. 40 487–40 503. [Online]. Available: https://procee...
2023
-
[35]
$\nablaˆ2$DFT: A universal quantum chemistry dataset of drug-like molecules and a benchmark for neural network potentials,
K. Khrabrov, A. Ber, A. Tsypin, K. Ushenin, E. Rumiantsev, A. Telepov, D. Protasov, I. Shenbin, A. M. Alekseev, M. Shirokikh, S. Nikolenko, E. Tutubalina, and A. Kadurin, “$\nablaˆ2$DFT: A universal quantum chemistry dataset of drug-like molecules and a benchmark for neural network potentials,” inThe Thirty-eight Conference on Neural Information Processin...
2024
-
[36]
A. S. Christensen and A. V . lilienfeld, “Revised MD17 dataset (rMD17),” 7 2020. [Online]. Available: https://doi.org/10.6084/m9. figshare.12672038.v3 3
work page doi:10.6084/m9 2020
-
[37]
H. Li, Z. Wang, N. Zou, M. Ye, R. Xu, X. Gong, W. Duan, and Y . Xu, “Deep-learning density functional theory hamiltonian for efficient ab initio electronic-structure calculation,”Nature Computational Science, vol. 2, no. 6, p. 367–377, Jun. 2022. [Online]. Available: http://dx.doi.org/10.1038/s43588-022-00265-6 3
-
[38]
The price of freedom: Exploring expressivity and runtime tradeoffs in equivariant tensor products,
Y . Xie, A. Daigavane, M. Kotak, and T. Smidt, “The price of freedom: Exploring expressivity and runtime tradeoffs in equivariant tensor products,” inF orty-second International Conference on Machine Learning, 2025. [Online]. Available: https://openreview.net/forum?id= EvIwwGYTLc 4
2025
-
[41]
Q. Gu, Z. Zhouyin, S. K. Pandey, P. Zhang, L. Zhang, and W. E, “Deep learning tight-binding approach for large-scale electronic simulations at finite temperatures with ab initio accuracy,”Nature Communications, vol. 15, no. 1, Aug. 2024. [Online]. Available: http://dx.doi.org/10.1038/s41467-024-51006-4 4
-
[42]
Physics-informed hamiltonian learning for large-scale optoelectronic property prediction,
M. Schwade, S. Zhang, F. V onhoff, F. P. Delgado, and D. A. Egger, “Physics-informed hamiltonian learning for large-scale optoelectronic property prediction,”Nature Communications, vol. 17, no. 1, Mar. 2026. [Online]. Available: http://dx.doi.org/10.1038/s41467-026-70865-7 4
-
[43]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, “LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales,”Comput. Phys. Commun.,...
-
[44]
DistMLIP: A distributed inference platform for machine learning interatomic potentials,
K. Han, B. Deng, A. B. Farimani, and G. Ceder, “DistMLIP: A distributed inference platform for machine learning interatomic potentials,” inThe F ourteenth International Conference on Learning Representations, 2026. [Online]. Available: https://openreview.net/ forum?id=4tasfBIPxp 4
2026
-
[45]
Understanding data movement in tightly coupled heterogeneous systems: A case study with the grace hopper superchip,
L. Fusco, M. Khalilov, M. Chrapek, G. Chukkapalli, T. Schulthess, and T. Hoefler, “Understanding data movement in tightly coupled heterogeneous systems: A case study with the grace hopper superchip,”
-
[47]
J. Ansel, E. Yang, H. He, N. Gimelshein, A. Jain, M. V oznesensky, B. Bao, P. Bell, D. Berard, E. Burovski, G. Chauhan, A. Chourdia, W. Constable, A. Desmaison, Z. DeVito, E. Ellison, W. Feng, J. Gong, M. Gschwind, B. Hirsh, S. Huang, K. Kalambarkar, L. Kirsch, M. Lazos, M. Lezcano, Y . Liang, J. Liang, Y . Lu, C. K. Luk, B. Maher, Y . Pan, C. Puhrsch, M....
-
[48]
M. Geiger and T. Smidt, “e3nn: Euclidean neural networks,” 2022. [Online]. Available: https://doi.org/10.48550/ARXIV .2207.09453 7
work page internal anchor Pith review doi:10.48550/arxiv 2022
-
[49]
Multilevelk-way partitioning scheme for irregular graphs,
G. Karypis and V . Kumar, “Multilevelk-way partitioning scheme for irregular graphs,”Journal of Parallel and Distributed Computing, vol. 48, no. 1, p. 96–129, Jan. 1998. [Online]. Available: http: //dx.doi.org/10.1006/jpdc.1997.1404 9
-
[50]
Device-scale atomistic modelling of phase-change memory materials,
Y . Zhou, W. Zhang, E. Ma, and V . L. Deringer, “Device-scale atomistic modelling of phase-change memory materials,”Nature Electronics, vol. 6, no. 10, p. 746–754, Sep. 2023. [Online]. Available: http://dx.doi.org/10.1038/s41928-023-01030-x 10, 11
-
[51]
Mobility calculation in disordered ws2-al2o3 stacks from first principles,
M. Dossena, B. Van Troeye, F. Ducry, J. Cao, A. Afzalian, G. Pourtois, and M. Luisier, “Mobility calculation in disordered ws2-al2o3 stacks from first principles,”npj 2D Materials and Applications, vol. 9, no. 1, Aug. 2025. [Online]. Available: http: //dx.doi.org/10.1038/s41699-025-00587-9 10
-
[52]
Equivariant representations for molecular hamiltonians and n-center atomic-scale properties,
J. Nigam, M. J. Willatt, and M. Ceriotti, “Equivariant representations for molecular hamiltonians and n-center atomic-scale properties,”The Journal of Chemical Physics, vol. 156, no. 1, Jan. 2022. [Online]. Available: http://dx.doi.org/10.1063/5.0072784 10
-
[53]
Silicon ribbonfet cmos at 6nm gate length,
G. Yeap, S. Lin, H. Shang, H. Lin, Y . Peng, M. Wang, P. Wang, C. Lin, K. Yu, W. Lee, H. Chen, D. Lin, B. Yang, C. Yeh, C. Chan, J. Kuo, C.-M. Liu, T. Chiu, M. Wen, T. Lee, C. Chang, R. Chen, P.-H. Huang, C. Hou, Y . Lin, F. Yang, J. Wang, S. Fung, R. Chen, C. Lee, T. Lee, W. Chang, D. Lee, C. Ting, T. Chang, H. Huang, H. Lin, C. Tseng, C. Chang, K. Huang...
-
[54]
Atomic origin of ultrafast resistance switching in nanoscale electrometallization cells,
N. Onofrio, D. Guzman, and A. Strachan, “Atomic origin of ultrafast resistance switching in nanoscale electrometallization cells,”Nature Materials, vol. 14, no. 4, p. 440–446, Mar. 2015. [Online]. Available: http://dx.doi.org/10.1038/nmat4221 11
-
[55]
An atomistic model of field-induced resistive switching in valence change memory,
M. Kaniselvan, M. Luisier, and M. Mladenovi ´c, “An atomistic model of field-induced resistive switching in valence change memory,”ACS Nano, vol. 17, no. 9, p. 8281–8292, Mar. 2023. [Online]. Available: http://dx.doi.org/10.1021/acsnano.2c12575 11
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.