CryoACE: An Atom-centric Framework for Accurate and Automated Model Building in Cryo-EM
Pith reviewed 2026-07-01 05:30 UTC · model grok-4.3
The pith
CryoACE builds atomic protein models from cryo-EM maps by sampling density directly at atom positions and applying local resolution guidance.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
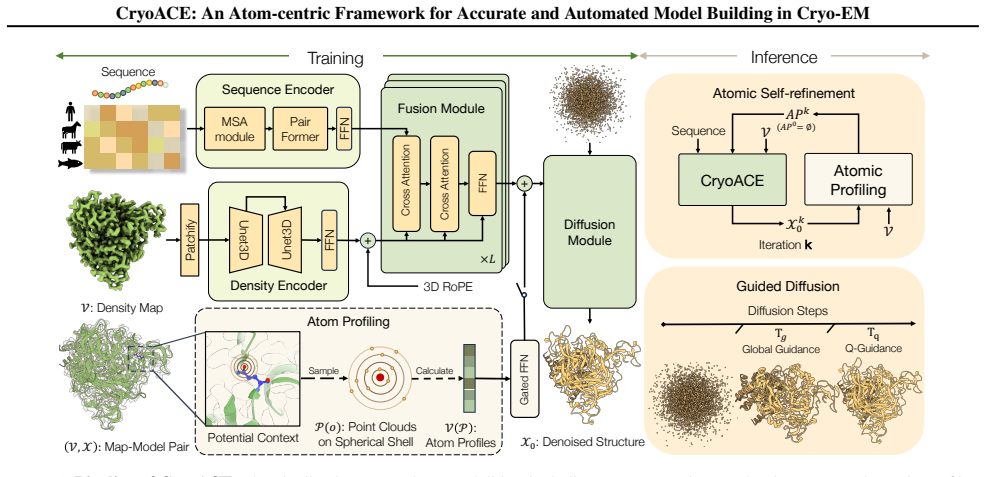
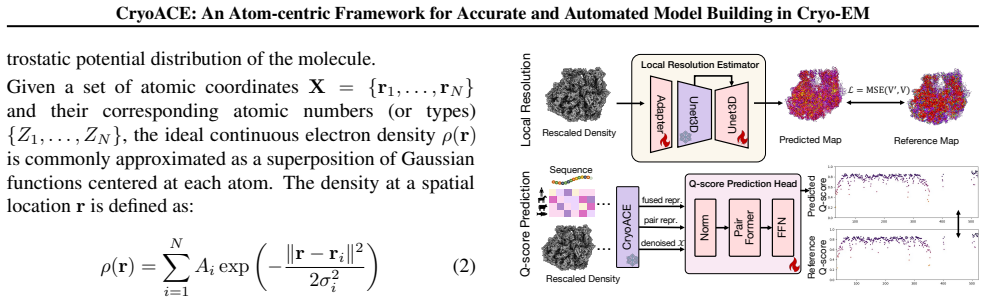
CryoACE reconstructs precise atomic graphs for homogeneous and heterogeneous structures through an atom-centric reconstruction paradigm in which density features are sampled directly at atomic coordinates and iteratively recycled to refine the model, replacing expensive voxel convolutions, together with a training-free guidance mechanism that leverages predicted local resolution priors to resolve dynamic ambiguity.
What carries the argument
The atom-centric reconstruction paradigm, which samples density features at atomic coordinates and recycles them iteratively to refine structures instead of using voxel convolutions.
If this is right
- Automated atomic model building becomes possible for heterogeneous maps without pre-built static structures.
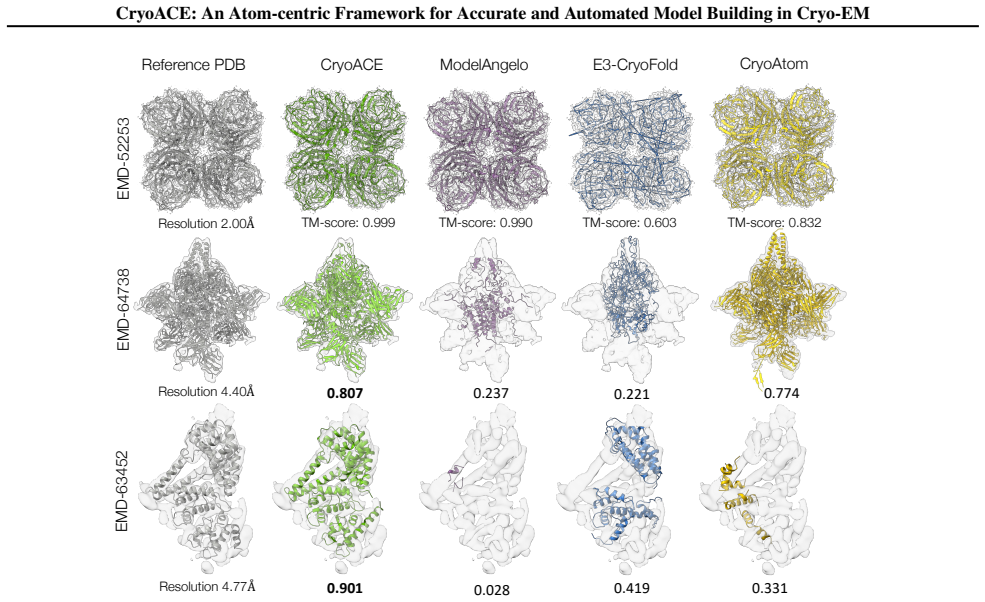
- The method produces higher accuracy than prior baselines on standard static test cases.
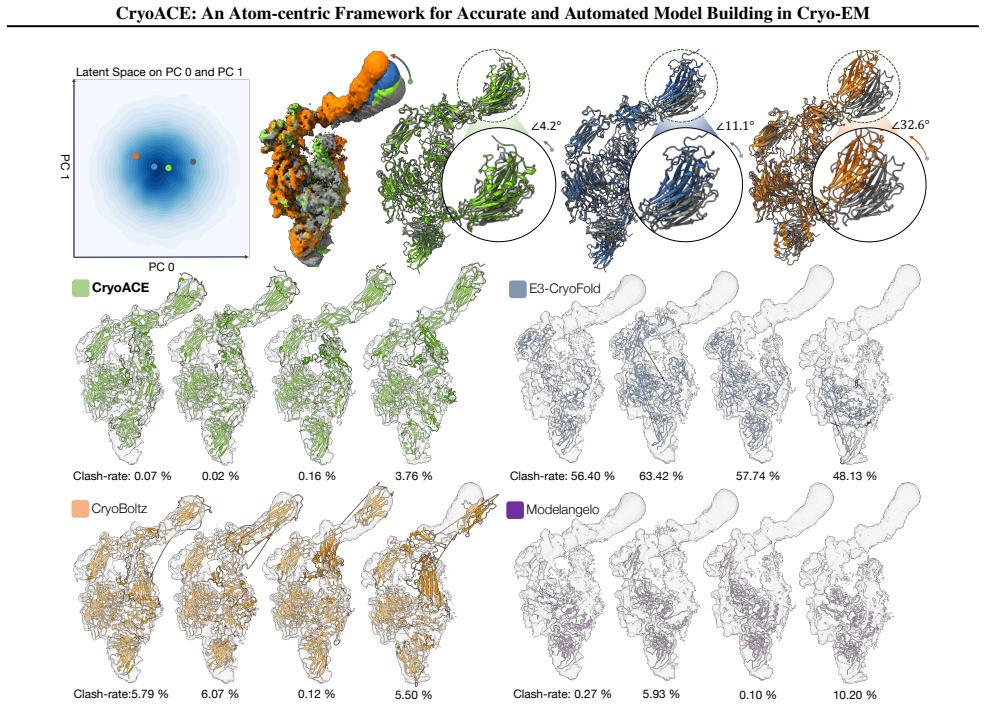
- Atomic-level dynamic conformations can be recovered directly from complex experimental datasets such as EMPIAR-10345.
- Physicochemical validity is maintained through the coordinate-based sampling loop and resolution guidance.
Where Pith is reading between the lines
- The same coordinate-sampling loop might be adapted to other density-based imaging methods where voxel grids are currently the default.
- If the local-resolution prior can be obtained cheaply, the framework could support iterative refinement during data collection rather than only after the fact.
- Success on EMPIAR-10345 suggests the approach may scale to larger assemblies where conformational heterogeneity has previously blocked atomic modeling.
Load-bearing premise
Sampling density at atomic coordinates together with local resolution guidance is enough to enforce chemical validity and remove conformational ambiguity in the maps.
What would settle it
Running CryoACE on a high-resolution map of a known static protein and finding that the output model contains bond lengths or angles outside accepted chemical ranges, or that it produces a single static model on EMPIAR-10345 when multiple distinct conformations are independently known to exist.
Figures


read the original abstract
Protein automodeling from cryo-EM density maps faces unique challenges in enforcing physicochemical validity and managing conformational heterogeneity. Current solvers are often limited to static predictions or require computationally intensive heuristic searches. We present CryoACE, an end-to-end framework that reconstructs precise atomic graphs for both homogeneous and heterogeneous structures. Our method features two key innovations: an atom-centric reconstruction paradigm, where density features are sampled directly at atomic coordinates and iteratively recycled to refine structures, replacing expensive voxel convolutions for efficient multimodal fusion; and a training-free guidance mechanism that leverages predicted local resolution priors to resolve dynamic ambiguity. Validated on a newly constructed high-quality dataset, CryoACE significantly outperforms existing baselines on static benchmarks and, for the first time, unveils atomic-level dynamic conformations on complex real-world datasets like EMPIAR-10345 without relying on pre-built static structures.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents CryoACE, an end-to-end atom-centric framework for automated atomic model building from cryo-EM density maps. It replaces voxel convolutions with direct sampling of density features at atomic coordinates, incorporates iterative recycling for refinement, and adds a training-free guidance step that uses predicted local resolution priors to address conformational heterogeneity. The abstract claims that the method significantly outperforms baselines on static benchmarks and, for the first time, recovers atomic-level dynamic conformations on real-world heterogeneous datasets such as EMPIAR-10345 without requiring pre-built static structures, all validated on a newly constructed high-quality dataset.
Significance. If the quantitative claims hold, the atom-centric sampling and training-free local-resolution guidance could provide an efficient alternative to existing voxel-based or heuristic cryo-EM modeling pipelines, particularly for heterogeneous maps. The absence of any reported metrics, however, prevents evaluation of whether these innovations actually deliver the stated gains in accuracy or physicochemical validity.
major comments (3)
- [Abstract] Abstract: the central claims that CryoACE 'significantly outperforms existing baselines on static benchmarks' and 'unveils atomic-level dynamic conformations on complex real-world datasets like EMPIAR-10345' are unsupported by any numerical results, tables, ablation studies, error bars, or dataset statistics. This evidentiary gap is load-bearing for the paper's primary contribution.
- [Abstract] Abstract: no description is supplied of the 'newly constructed high-quality dataset,' its size, composition, resolution range, or how it differs from public benchmarks, nor are any specific performance numbers (e.g., RMSD, completeness, or local-resolution correlation) reported for either the static or dynamic cases. Without these details the outperformance and novelty assertions cannot be assessed.
- [Abstract] Abstract: the atom-centric sampling and local-resolution guidance are asserted to enforce 'physicochemical validity' and resolve 'conformational ambiguity,' yet the abstract supplies no concrete mechanism, loss term, or validation metric showing that these steps produce stereochemically valid models or disambiguate states better than existing methods.
minor comments (1)
- [Abstract] The abstract would be clearer if it included at least one key quantitative result (e.g., average RMSD or success rate) to anchor the performance claims.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments on the manuscript. We address each major comment below and agree that the abstract can be strengthened with additional concrete details drawn from the full paper to better support the claims.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claims that CryoACE 'significantly outperforms existing baselines on static benchmarks' and 'unveils atomic-level dynamic conformations on complex real-world datasets like EMPIAR-10345' are unsupported by any numerical results, tables, ablation studies, error bars, or dataset statistics. This evidentiary gap is load-bearing for the paper's primary contribution.
Authors: The full manuscript reports these quantitative results, including tables with RMSD, completeness, and local-resolution metrics, ablation studies, and error bars in the Results section. The abstract was intentionally concise, but we agree it would benefit from key numerical highlights and will revise it to include representative performance numbers and dataset statistics. revision: yes
-
Referee: [Abstract] Abstract: no description is supplied of the 'newly constructed high-quality dataset,' its size, composition, resolution range, or how it differs from public benchmarks, nor are any specific performance numbers (e.g., RMSD, completeness, or local-resolution correlation) reported for either the static or dynamic cases. Without these details the outperformance and novelty assertions cannot be assessed.
Authors: The dataset construction, size, composition, resolution range, and differences from public benchmarks are detailed in the Methods section. We will add a brief summary of these elements and example performance numbers to the abstract. revision: yes
-
Referee: [Abstract] Abstract: the atom-centric sampling and local-resolution guidance are asserted to enforce 'physicochemical validity' and resolve 'conformational ambiguity,' yet the abstract supplies no concrete mechanism, loss term, or validation metric showing that these steps produce stereochemically valid models or disambiguate states better than existing methods.
Authors: The atom-centric sampling (replacing voxel convolutions with direct coordinate sampling and iterative recycling) and training-free local-resolution guidance, including the associated loss terms and stereochemical validation metrics, are described in Sections 3.2 and 3.3. We will revise the abstract to include a concise reference to these mechanisms. revision: yes
Circularity Check
No significant circularity; derivation self-contained against external benchmarks
full rationale
The abstract and available description introduce an atom-centric sampling paradigm and training-free local-resolution guidance as methodological choices, but present no equations, fitted parameters renamed as predictions, self-citation chains, or uniqueness theorems that reduce the claimed outputs to the inputs by construction. Performance claims are tied to external benchmarks (static sets and EMPIAR-10345) rather than internal recycling of the same quantities. This is the normal case of an empirical method whose validity rests on independent validation rather than definitional equivalence.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Proceedings of the AAAI Conference on Artificial Intelligence , volume=
3d-rpe: Enhancing long-context modeling through 3d rotary position encoding , author=. Proceedings of the AAAI Conference on Artificial Intelligence , volume=
-
[2]
International conference on medical image computing and computer-assisted intervention , pages=
3D U-Net: learning dense volumetric segmentation from sparse annotation , author=. International conference on medical image computing and computer-assisted intervention , pages=. 2016 , organization=
2016
-
[3]
Punjani, Ali and Rubinstein, John L. and Fleet, David J. and Brubaker, Marcus A. , title=. Nature Methods , year=. doi:10.1038/nmeth.4169 , url=
-
[4]
Nature , volume=
Automated model building and protein identification in cryo-EM maps , author=. Nature , volume=. 2024 , publisher=
2024
-
[5]
Nature Structural & Molecular Biology , pages=
CryoAtom improves model building for cryo-EM , author=. Nature Structural & Molecular Biology , pages=. 2025 , publisher=
2025
-
[6]
Nature Machine Intelligence , pages=
End-to-end Cryo-EM complex structure determination with high accuracy and ultra-fast speed , author=. Nature Machine Intelligence , pages=. 2025 , publisher=
2025
-
[7]
arXiv preprint arXiv:2506.04490 , year=
Multiscale guidance of AlphaFold3 with heterogeneous cryo-EM data , author=. arXiv preprint arXiv:2506.04490 , year=
-
[8]
Nature methods , volume=
CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks , author=. Nature methods , volume=. 2021 , publisher=
2021
-
[9]
The Buccaneer software for automated model building. 1. Tracing protein chains , author=. Biological Crystallography , volume=. 2006 , publisher=
2006
-
[10]
Nature communications , volume=
De novo main-chain modeling for EM maps using MAINMAST , author=. Nature communications , volume=. 2018 , publisher=
2018
-
[11]
Nature methods , volume=
A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps , author=. Nature methods , volume=. 2018 , publisher=
2018
-
[12]
Structure , volume=
Constructing and validating initial C models from subnanometer resolution density maps with pathwalking , author=. Structure , volume=. 2012 , publisher=
2012
-
[13]
Journal of structural biology , volume=
De Novo modeling in cryo-EM density maps with Pathwalking , author=. Journal of structural biology , volume=. 2016 , publisher=
2016
-
[14]
Nature methods , volume=
De novo protein structure determination from near-atomic-resolution cryo-EM maps , author=. Nature methods , volume=. 2015 , publisher=
2015
-
[15]
Structure , volume=
EM-fold: De novo folding of -helical proteins guided by intermediate-resolution electron microscopy density maps , author=. Structure , volume=. 2009 , publisher=
2009
-
[16]
Biological crystallography , volume=
Coot: model-building tools for molecular graphics , author=. Biological crystallography , volume=. 2004 , publisher=
2004
-
[17]
IEEE/ACM transactions on computational biology and bioinformatics , volume=
Intensity-based skeletonization of CryoEM gray-scale images using a true segmentation-free algorithm , author=. IEEE/ACM transactions on computational biology and bioinformatics , volume=. 2013 , publisher=
2013
-
[18]
BioRxiv , pages=
Boltz-1 democratizing biomolecular interaction modeling , author=. BioRxiv , pages=
-
[19]
BioRxiv , year=
Boltz-2: Towards accurate and efficient binding affinity prediction , author=. BioRxiv , year=
-
[20]
Nature , volume=
Improved protein structure prediction using potentials from deep learning , author=. Nature , volume=. 2020 , publisher=
2020
-
[21]
nature , volume=
Highly accurate protein structure prediction with AlphaFold , author=. nature , volume=. 2021 , publisher=
2021
-
[22]
Nature , volume=
Accurate structure prediction of biomolecular interactions with AlphaFold 3 , author=. Nature , volume=. 2024 , publisher=
2024
-
[23]
Nature methods , volume=
OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization , author=. Nature methods , volume=. 2024 , publisher=
2024
-
[24]
arXiv preprint arXiv:2512.24354 , year=
SeedFold: Scaling Biomolecular Structure Prediction , author=. arXiv preprint arXiv:2512.24354 , year=
-
[25]
Science , volume=
Accurate prediction of protein structures and interactions using a three-track neural network , author=. Science , volume=. 2021 , publisher=
2021
-
[26]
Nature methods , volume=
Accurate prediction of protein--nucleic acid complexes using RoseTTAFoldNA , author=. Nature methods , volume=. 2024 , publisher=
2024
-
[27]
Advances in neural information processing systems , volume=
Language models enable zero-shot prediction of the effects of mutations on protein function , author=. Advances in neural information processing systems , volume=
-
[28]
Science , volume=
Evolutionary-scale prediction of atomic-level protein structure with a language model , author=. Science , volume=. 2023 , publisher=
2023
-
[29]
Science , volume=
Simulating 500 million years of evolution with a language model , author=. Science , volume=. 2025 , publisher=
2025
-
[30]
Nature reviews molecular cell biology , volume=
Advances in protein structure prediction and design , author=. Nature reviews molecular cell biology , volume=. 2019 , publisher=
2019
-
[31]
Frontiers in Bioinformatics , volume=
Protein structure prediction in the era of AI: Challenges and limitations when applying to in silico force spectroscopy , author=. Frontiers in Bioinformatics , volume=. 2022 , publisher=
2022
-
[32]
Prediction of Protein Secondary Structure , pages=
Beyond AlphaFold2: the impact of AI for the further improvement of protein structure prediction , author=. Prediction of Protein Secondary Structure , pages=. 2024 , publisher=
2024
-
[33]
Frontiers in Pharmacology , volume=
Protein structure prediction via deep learning: an in-depth review , author=. Frontiers in Pharmacology , volume=. 2025 , publisher=
2025
-
[34]
Scientific reports , volume=
Deep learning to predict protein backbone structure from high-resolution cryo-EM density maps , author=. Scientific reports , volume=. 2020 , publisher=
2020
-
[35]
Nature methods , volume=
CR-I-TASSER: assemble protein structures from cryo-EM density maps using deep convolutional neural networks , author=. Nature methods , volume=. 2022 , publisher=
2022
-
[36]
Proceedings of the National Academy of Sciences , volume=
DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes , author=. Proceedings of the National Academy of Sciences , volume=. 2021 , publisher=
2021
-
[37]
Nature Communications , volume=
De novo atomic protein structure modeling for cryoEM density maps using 3D transformer and HMM , author=. Nature Communications , volume=. 2024 , publisher=
2024
-
[38]
Nature Communications , volume=
Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly , author=. Nature Communications , volume=. 2022 , publisher=
2022
-
[39]
Science , volume=
Protein structure relationships revealed by mutational analysis , author=. Science , volume=. 1964 , publisher=
1964
-
[40]
Protein Engineering, Design and Selection , volume=
Coordinated amino acid changes in homologous protein families , author=. Protein Engineering, Design and Selection , volume=. 1988 , publisher=
1988
-
[41]
Proteins: Structure, Function, and Bioinformatics , volume=
Correlated mutations and residue contacts in proteins , author=. Proteins: Structure, Function, and Bioinformatics , volume=. 1994 , publisher=
1994
-
[42]
Nature Biotechnology , volume=
Single-sequence protein structure prediction using a language model and deep learning , author=. Nature Biotechnology , volume=. 2022 , publisher=
2022
-
[43]
homology
An introduction to sequence similarity (“homology”) searching , author=. Current protocols in bioinformatics , volume=. 2013 , publisher=
2013
-
[44]
Proceedings of the National Academy of Sciences , volume=
Unexpected features of the dark proteome , author=. Proceedings of the National Academy of Sciences , volume=. 2015 , publisher=
2015
-
[45]
Nature methods , volume=
Residue-wise local quality estimation for protein models from cryo-EM maps , author=. Nature methods , volume=. 2022 , publisher=
2022
-
[46]
Biological crystallography , volume=
MolProbity: all-atom structure validation for macromolecular crystallography , author=. Biological crystallography , volume=. 2010 , publisher=
2010
-
[47]
Nature methods , volume=
Protein secondary structure detection in intermediate-resolution cryo-EM maps using deep learning , author=. Nature methods , volume=. 2019 , publisher=
2019
-
[48]
Nature Methods , volume=
CryoREAD: de novo structure modeling for nucleic acids in cryo-EM maps using deep learning , author=. Nature Methods , volume=. 2023 , publisher=
2023
-
[49]
Nature communications , volume=
NuFold: end-to-end approach for RNA tertiary structure prediction with flexible nucleobase center representation , author=. Nature communications , volume=. 2025 , publisher=
2025
-
[50]
Nature Methods , volume=
DeepMainmast: integrated protocol of protein structure modeling for cryo-EM with deep learning and structure prediction , author=. Nature Methods , volume=. 2024 , publisher=
2024
-
[51]
Microscopy and Microanalysis , volume=
DMcloud: Macromolecular Structure Modeling with Local Structure Fitting for Medium to Low Resolution Cryo-EM Maps , author=. Microscopy and Microanalysis , volume=. 2025 , publisher=
2025
-
[52]
Nature Methods , volume=
DiffModeler: large macromolecular structure modeling for cryo-EM maps using a diffusion model , author=. Nature Methods , volume=. 2024 , publisher=
2024
-
[53]
Nucleic acids research , volume=
EMDataBank unified data resource for 3DEM , author=. Nucleic acids research , volume=. 2016 , publisher=
2016
-
[54]
Nucleic acids research , volume=
RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences , author=. Nucleic acids research , volume=. 2021 , publisher=
2021
-
[55]
Scientific Data , volume=
Cryo2structdata: A large labeled cryo-em density map dataset for ai-based modeling of protein structures , author=. Scientific Data , volume=. 2024 , publisher=
2024
-
[56]
Nature methods , volume=
Quantifying the local resolution of cryo-EM density maps , author=. Nature methods , volume=. 2014 , publisher=
2014
-
[57]
Nature Communications , volume=
ARMH2 is a cytosolic component of CatSper crucial for sperm function , author=. Nature Communications , volume=. 2025 , publisher=
2025
-
[58]
and Vriends, M
Moran, E. and Vriends, M. and Calvelo, M. and Hansen, T. and Pickles, I. and Xin, X. and Biezeno, M. and Armstrong, Z. and Ferras, M. and Li, L. and Lilley, A. and Harvey, R. and Schr. Influenza Neuraminidase in complex with N-Acyl Oseltamivir inhibitor , year =
-
[59]
2025 , publisher =
Zhai H, Deng J, Yu W , title =. 2025 , publisher =
2025
-
[60]
and Lo, Wan-Yen and Dollar, Piotr and Girshick, Ross , title =
Kirillov, Alexander and Mintun, Eric and Ravi, Nikhila and Mao, Hanzi and Rolland, Chloe and Gustafson, Laura and Xiao, Tete and Whitehead, Spencer and Berg, Alexander C. and Lo, Wan-Yen and Dollar, Piotr and Girshick, Ross , title =. Proceedings of the IEEE/CVF International Conference on Computer Vision (ICCV) , month =. 2023 , pages =
2023
-
[61]
Grounding DINO: Marrying DINO with Grounded Pre-training for Open-Set Object Detection
Liu, Shilong and Zeng, Zhaoyang and Ren, Tianhe and Li, Feng and Zhang, Hao and Yang, Jie and Jiang, Qing and Li, Chunyuan and Yang, Jianwei and Su, Hang and Zhu, Jun and Zhang, Lei. Grounding DINO: Marrying DINO with Grounded Pre-training for Open-Set Object Detection. Computer Vision -- ECCV 2024. 2025
2024
-
[62]
Cell , volume=
Cryo-EM reveals integrin-mediated TGF- activation without release from latent TGF- , author=. Cell , volume=. 2020 , publisher=
2020
-
[63]
IUCrJ , volume=
Continuous flexibility analysis of SARS-CoV-2 spike prefusion structures , author=. IUCrJ , volume=. 2020 , publisher=
2020
-
[64]
Nature methods , volume=
EMPIAR: a public archive for raw electron microscopy image data , author=. Nature methods , volume=. 2016 , publisher=
2016
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.