High-performance parallel implementation of high-order coupled-cluster theories
Pith reviewed 2026-07-02 04:21 UTC · model grok-4.3
The pith
Distributed parallel code makes high-order coupled-cluster calculations feasible for molecules with 100 correlated electrons.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors implement spin-restricted and unrestricted high-order coupled-cluster methods that store high-order amplitudes in compact triangular form and distribute them across MPI ranks while overlapping communication with computation through nonblocking transfers, thereby achieving near-ideal strong scaling that enables CCSDT(Q) calculations on approximately 100 correlated electrons in 450 orbitals and CCSDTQ calculations on approximately 50 correlated electrons in 115 orbitals.
What carries the argument
Compact triangular storage of highest-order amplitude tensors combined with nonblocking MPI data transfers across distributed ranks.
If this is right
- CCSDT(Q) calculations become practical for systems with approximately 100 correlated electrons in 450 orbitals.
- CCSDTQ calculations become practical for systems with approximately 50 correlated electrons in 115 orbitals.
- Canonical high-order CC can now be applied to pi-stacked noncovalent dimers, CO dissociation in Cr(CO)6, and the Cope rearrangement of semibullvalene.
- Single-node shared-memory versions deliver near-ideal thread scaling up to 90 cores.
Where Pith is reading between the lines
- The same storage and communication pattern could be tested on other tensor-network methods whose scaling is dominated by high-order contractions.
- Adoption of the open-source code could enable direct comparison of high-order CC results against lower-scaling approximations on identical molecular geometries.
- The demonstrated node counts suggest that further extension to 64 nodes would be a straightforward next measurement to confirm continued linear behavior.
Load-bearing premise
Compact triangular storage of high-order amplitudes together with nonblocking MPI transfers produces negligible overhead and memory fragmentation on the benchmark molecules and orbital counts tested.
What would settle it
A CCSDT(Q) run on a 100-electron system in 450 orbitals that fails to finish in the time predicted by linear extrapolation from smaller node counts or shows clear deviation from ideal scaling on 32 nodes would falsify the performance claim.
Figures

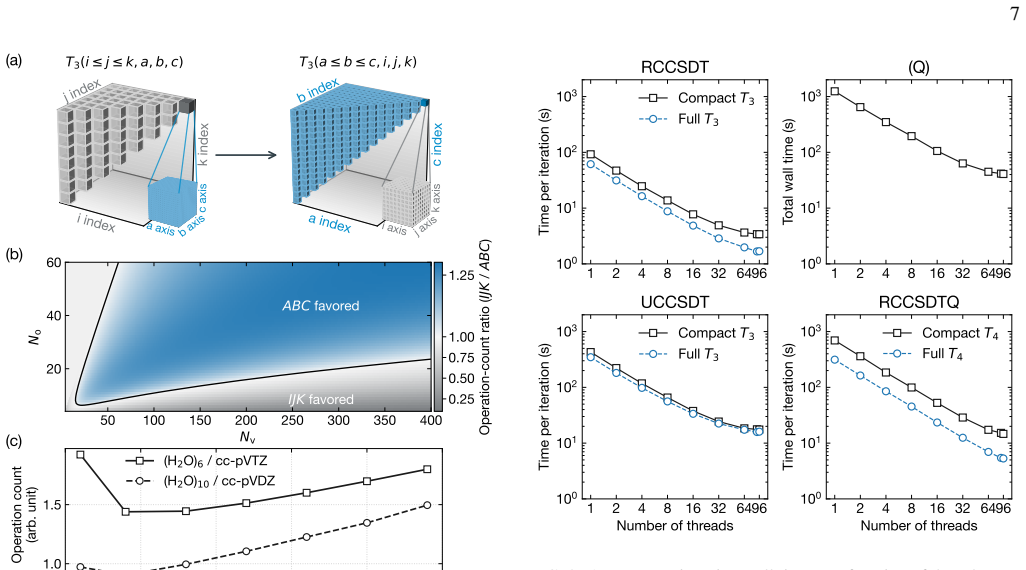

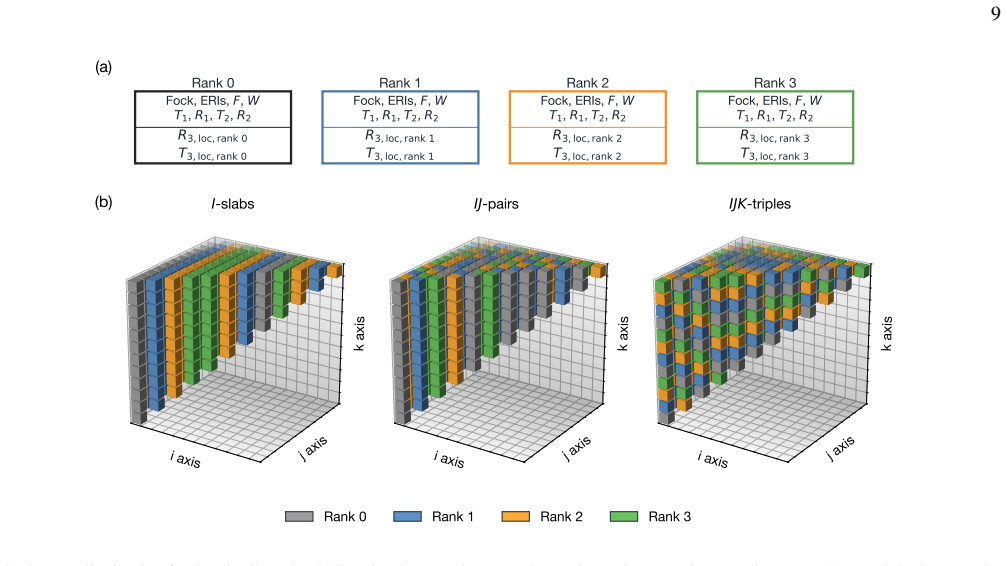

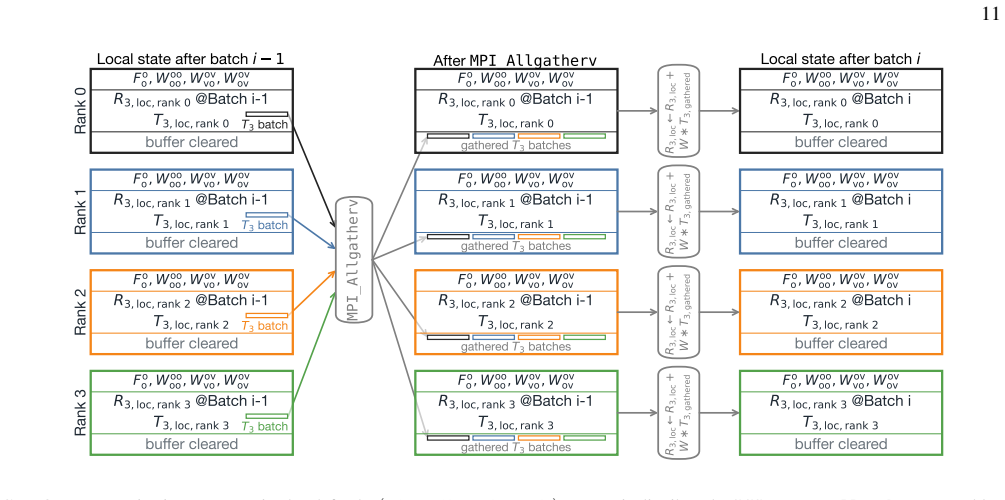
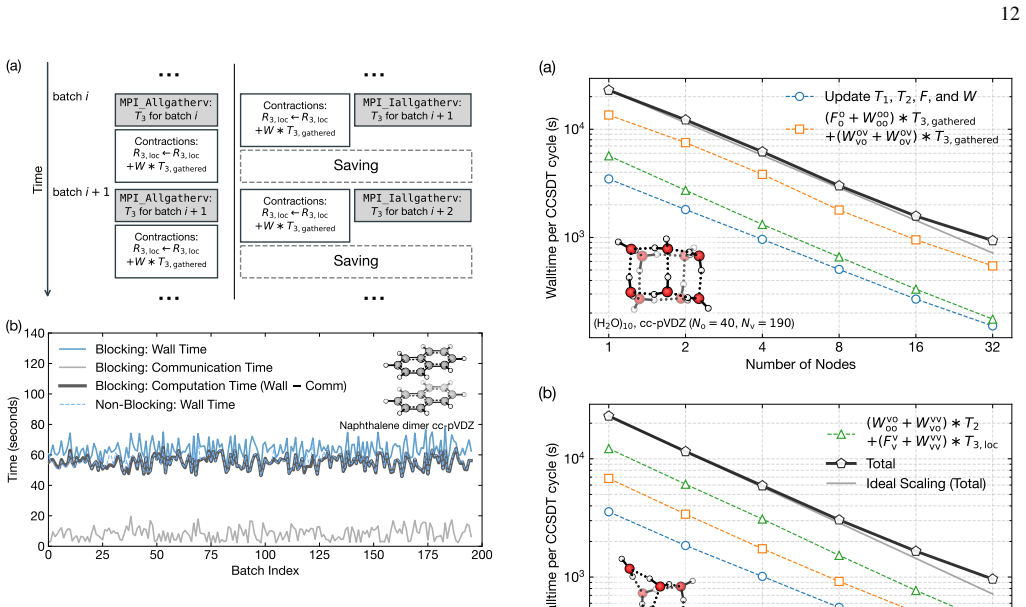
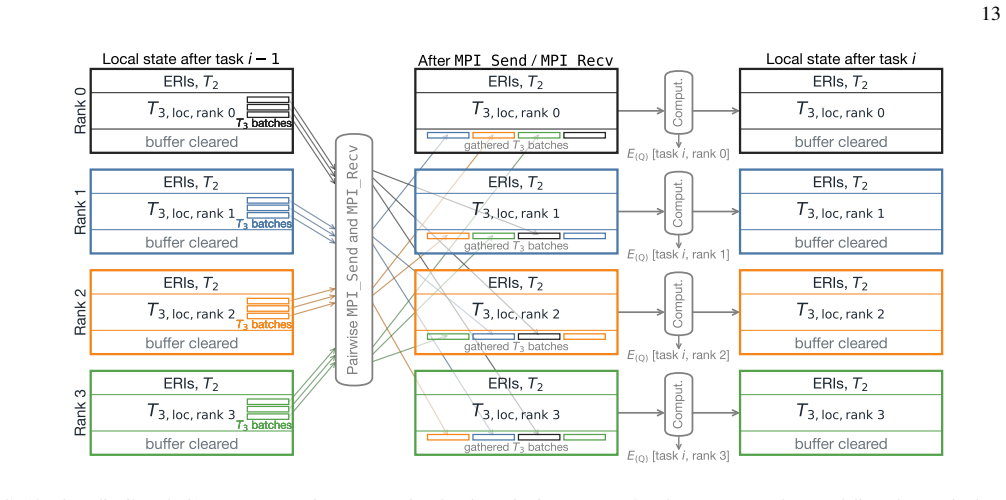
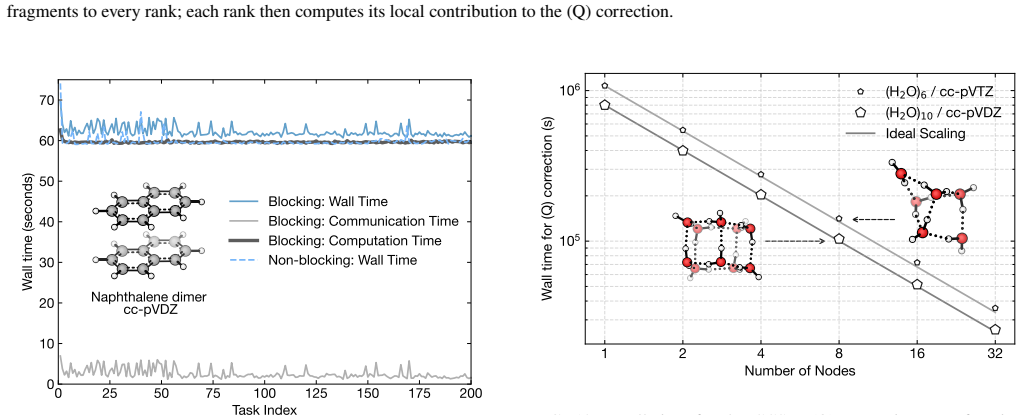
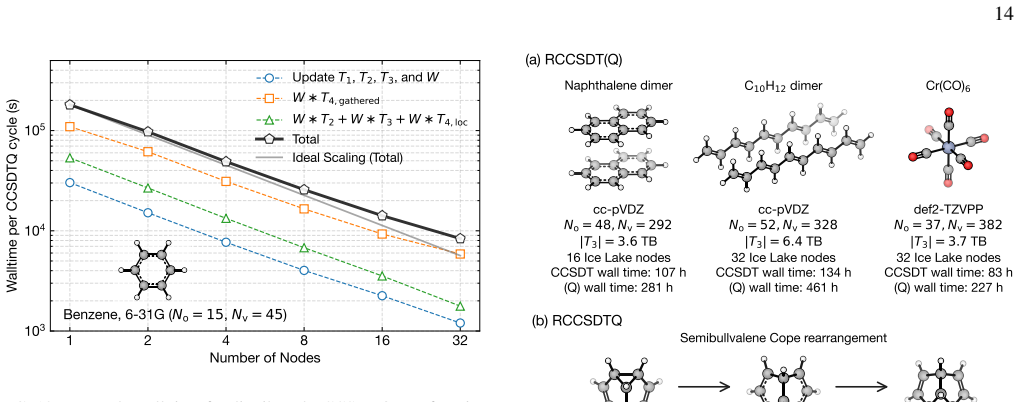
read the original abstract
High-order coupled-cluster theories with iterative triples (CCSDT), perturbative quadruples [CCSDT(Q)], and iterative quadruples (CCSDTQ) provide benchmark-quality correlation energies, but their steep computational scalings, $O(N^8), O(N^9)$, and $O(N^{10})$, together with the large memory requirements of high-order amplitude tensors, have historically limited their application to small molecules. In this work, we develop efficient open-source implementations of spin-restricted CCSDT (RCCSDT), RCCSDT(Q), RCCSDTQ, and spin-unrestricted CCSDT (UCCSDT) within the PySCF package. The shared-memory implementation combines compact triangular storage of the highest-order amplitude tensors with the multithreaded tensor contraction backend pytblis, enabling efficient use of modern many-core CPU architectures. This design delivers near-ideal thread scaling up to 90 cores and achieves wall times shorter than or comparable to existing single-node implementations for representative benchmark molecules. We further extend RCCSDT, RCCSDT(Q), and RCCSDTQ to distributed-memory architectures using MPI-based algorithms. By distributing compact high-order amplitudes across MPI ranks and overlapping communication with computation through nonblocking data transfers, the distributed implementation achieves near-ideal strong scaling on up to 32 nodes, corresponding to approximately 3,000 CPU cores. These developments substantially extend the practical reach of canonical high-order CC theory, enabling CCSDT(Q) calculations with approximately 100 correlated electrons in 450 orbitals and CCSDTQ calculations with approximately 50 correlated electrons in 115 orbitals. Applications to $\pi$-stacked noncovalent dimers, the CO dissociation energy of Cr(CO)$_6$, and the Cope rearrangement of semibullvalene demonstrate that canonical high-order CC benchmarks are now feasible for chemically realistic molecular systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents open-source implementations of spin-restricted and unrestricted high-order coupled-cluster methods (RCCSDT, RCCSDT(Q), RCCSDTQ, UCCSDT) within PySCF. It combines compact triangular storage of high-order amplitudes with the pytblis tensor contraction library for shared-memory parallelism, achieving near-ideal thread scaling to 90 cores. The work further extends these methods to distributed memory via MPI, distributing amplitudes and using nonblocking transfers to overlap communication with computation, claiming near-ideal strong scaling to 32 nodes (~3000 cores). This enables CCSDT(Q) calculations with ~100 correlated electrons in 450 orbitals and CCSDTQ with ~50 electrons in 115 orbitals, with applications to π-stacked dimers, Cr(CO)6 dissociation, and semibullvalene Cope rearrangement.
Significance. If the performance claims hold, the work meaningfully extends the practical applicability of canonical high-order CC theory to chemically realistic systems that were previously inaccessible due to scaling and memory limits. The open-source release in PySCF and the focus on both shared- and distributed-memory architectures represent concrete advances for the field.
major comments (1)
- [Abstract] Abstract: The central performance claims rest on the assertion that compact triangular storage of high-order amplitudes combined with nonblocking MPI transfers incurs negligible overhead relative to pytblis contractions. However, the text provides no per-phase timing tables, memory high-water-mark data, or strong-scaling efficiency metrics that would confirm this assumption holds at the largest scales (e.g., 450 orbitals for CCSDT(Q)).
minor comments (1)
- The abstract mentions specific molecule sizes and orbital counts but does not reference the corresponding benchmark tables or figures that would allow direct verification of the reported wall times and scaling.
Simulated Author's Rebuttal
We thank the referee for their careful review and constructive comment. We address the single major comment below and will revise the manuscript to incorporate additional performance data.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central performance claims rest on the assertion that compact triangular storage of high-order amplitudes combined with nonblocking MPI transfers incurs negligible overhead relative to pytblis contractions. However, the text provides no per-phase timing tables, memory high-water-mark data, or strong-scaling efficiency metrics that would confirm this assumption holds at the largest scales (e.g., 450 orbitals for CCSDT(Q)).
Authors: We agree that the manuscript would be strengthened by more granular performance metrics at the largest scales. While the current text includes overall wall-time comparisons, thread-scaling plots to 90 cores, and MPI strong-scaling data to 32 nodes, it does not provide explicit per-phase breakdowns (e.g., contraction vs. communication time), memory high-water marks, or numerical strong-scaling efficiencies specifically for the ~100-electron/450-orbital CCSDT(Q) case. In the revised manuscript we will add a dedicated table (or supplementary figure) reporting these quantities for the largest calculations, confirming that storage and nonblocking MPI overhead remains negligible relative to the pytblis contractions. revision: yes
Circularity Check
No circularity: pure implementation and performance paper
full rationale
This is a pure implementation and performance paper with no derivations, fitted parameters, predictions, or self-referential theoretical claims. The reported scaling results and enabled system sizes are direct empirical outcomes from benchmarks, not reductions by construction to inputs or self-citations. The paper is self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard assumptions of spin-restricted and unrestricted coupled-cluster theory remain valid for the tested molecular systems.
Reference graph
Works this paper leans on
-
[1]
Coupled-cluster theory in quantum chemistry , author =. Rev. Mod. Phys. , volume =. 2007 , month =. doi:10.1103/RevModPhys.79.291 , url =
-
[2]
2009 , publisher=
Many-body methods in chemistry and physics: MBPT and coupled-cluster theory , author=. 2009 , publisher=
2009
-
[3]
An introduction to coupled cluster theory for computational chemists , author=. Rev. Comput. Chem. , volume=. 2007 , url=
2007
-
[4]
The full
Noga, Jozef and Bartlett, Rodney J , journal=. The full. 1987 , publisher=
1987
-
[5]
Erratum: The full
Noga, J and Bartlett, RJ , journal=. Erratum: The full. 1988 , publisher=
1988
-
[6]
A new implementation of the full
Scuseria, Gustavo E and Schaefer. A new implementation of the full. Chem. Phys. Lett. , volume=. 1988 , publisher=
1988
-
[7]
The coupled-cluster single, double, triple, and quadruple excitation method , author=. J. Chem. Phys. , volume=. 1992 , publisher=
1992
-
[8]
Coupled-cluster techniques for computational chemistry: The
Matthews, Devin A and Cheng, Lan and Harding, Michael E and Lipparini, Filippo and Stopkowicz, Stella and Jagau, Thomas-C and Szalay, P. Coupled-cluster techniques for computational chemistry: The. J. Chem. Phys. , volume=. 2020 , publisher=
2020
-
[9]
J. F. Stanton and J. Gauss and L. Cheng and M. E. Harding and F. Lipparini and D. A. Matthews and P. G. Szalay and S. Stopkowicz , Note =
-
[10]
Parallel calculation of
Prochnow, Eric and Harding, Michael E and Gauss, Jürgen , journal=. Parallel calculation of. 2010 , publisher=
2010
-
[11]
A massively parallel implementation of the
Datta, Dipayan and Gordon, Mark S , journal=. A massively parallel implementation of the. 2021 , publisher=
2021
-
[12]
Massive-parallel implementation of the resolution-of-identity coupled-cluster approaches in the numeric atom-centered orbital framework for molecular systems , author=. J. Chem. Theory Comput. , volume=. 2019 , publisher=
2019
-
[13]
Massively parallel implementation of explicitly correlated coupled-cluster singles and doubles using
Peng, Chong and Calvin, Justus A and Pavosevic, Fabijan and Zhang, Jinmei and Valeev, Edward F , journal=. Massively parallel implementation of explicitly correlated coupled-cluster singles and doubles using. 2016 , publisher=
2016
-
[14]
Enabling accurate and large-scale explicitly correlated
Lad. Enabling accurate and large-scale explicitly correlated. J. Chem. Theory Comput. , volume=. 2025 , publisher=
2025
-
[15]
2013 IEEE 27th International Symposium on Parallel and Distributed Processing , pages=
Cyclops tensor framework: Reducing communication and eliminating load imbalance in massively parallel contractions , author=. 2013 IEEE 27th International Symposium on Parallel and Distributed Processing , pages=. 2013 , organization=
2013
-
[16]
A massively parallel tensor contraction framework for coupled-cluster computations , author=. J. Parallel Distrib. Comput. , volume=. 2014 , publisher=
2014
-
[17]
2020 , publisher=
Apra, Edoardo and Bylaska, Eric J and De Jong, Wibe A and Govind, Niranjan and Kowalski, Karol and Straatsma, Tjerk P and Valiev, Marat and van Dam, Hubertus JJ and Alexeev, Yuri and Anchell, James and others , journal=. 2020 , publisher=
2020
-
[18]
Tensor contraction engine: Abstraction and automated parallel implementation of configuration-interaction, coupled-cluster, and many-body perturbation theories , author=. J. Phys. Chem. A , volume=. 2003 , publisher=
2003
-
[19]
Synthesis of high-performance parallel programs for a class of ab initio quantum chemistry models , author=. Proc. IEEE , volume=. 2005 , publisher=
2005
-
[20]
1996 , publisher=
Nieplocha, Jaroslaw and Harrison, Robert J and Littlefield, Richard J , journal=. 1996 , publisher=
1996
-
[21]
Code generation in
Lechner, Marvin H and Papadopoulos, Anastasios and Sivalingam, Kantharuban and Auer, Alexander A and Koslowski, Axel and Becker, Ute and Wennmohs, Frank and Neese, Frank , journal=. Code generation in. 2024 , publisher=
2024
-
[22]
Software for the frontiers of quantum chemistry: An overview of developments in the
Epifanovsky, Evgeny and Gilbert, Andrew TB and Feng, Xintian and Lee, Joonho and Mao, Yuezhi and Mardirossian, Narbe and Pokhilko, Pavel and White, Alec F and Coons, Marc P and Dempwolff, Adrian L and others , journal=. Software for the frontiers of quantum chemistry: An overview of developments in the. 2021 , publisher=
2021
-
[23]
Frozen Natural Orbitals-Based Coupled-Cluster Singles, Doubles, and (full) Triples---A Computational Study , author=. Chem. Asian J. , volume=. 2025 , publisher=
2025
-
[24]
Emiliano and Piecuch, Piotr , howpublished=
Gururangan, Karthik and Deustua, J. Emiliano and Piecuch, Piotr , howpublished=. 2026 , url=
2026
-
[25]
Folkestad, Sarai Dery and Kj. J. Chem. Phys. , volume=. 2026 , publisher=
2026
-
[26]
Overview of developments in the
Mester, D. Overview of developments in the. J. Phys. Chem. A , volume=. 2025 , publisher=
2025
-
[27]
Higher excitations in coupled-cluster theory , author=. J. Chem. Phys. , volume=. 2001 , publisher=
2001
-
[28]
Approximate treatment of higher excitations in coupled-cluster theory , author=. J. Chem. Phys. , volume=. 2005 , publisher=
2005
-
[29]
Coupled-cluster methods including noniterative corrections for quadruple excitations , author=. J. Chem. Phys. , volume=. 2005 , publisher=
2005
-
[30]
Approximate treatment of higher excitations in coupled-cluster theory
K. Approximate treatment of higher excitations in coupled-cluster theory. J. Chem. Phys. , volume=. 2008 , publisher=
2008
-
[31]
Generating Coupled Cluster Code for Modern Distributed-Memory Tensor Software , author=. J. Chem. Theory Comput. , volume=. 2025 , publisher=
2025
-
[32]
Implementation of relativistic coupled cluster theory for massively parallel GPU-accelerated computing architectures , author=. J. Chem. Theory Comput. , volume=. 2021 , publisher=
2021
-
[33]
Penguin: A
Hillers-Bendtsen, Andreas Erbs and Johansen, Magnus Bukhave and von Buchwald, Theo Juncker and Dunweber, Phillip Gustav Iuel Lunoe and Olsen, Lars Henrik and Rask, Laust and Junker, Georg Ingvartsen and Knudsen, Rasmine Maria Hansen and Mikkelsen, Kurt V , journal=. Penguin: A. 2025 , publisher=
2025
-
[34]
Accelerating CCSD (T) on Graphical Processing Units (GPUs) , author=. J. Phys. Chem. A , volume=. 2026 , publisher=
2026
-
[35]
Accelerating the convergence of higher-order coupled cluster methods , author=. J. Chem. Phys. , volume=. 2015 , publisher=
2015
-
[36]
Accelerating the convergence of higher-order coupled-cluster methods
Matthews, Devin A , journal=. Accelerating the convergence of higher-order coupled-cluster methods. 2020 , publisher=
2020
-
[37]
Classical computational simulation of the FeMo-cofactor model to chemical accuracy and its implications , author=. arXiv:2601.04621 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[38]
Periodic local coupled-cluster theory for insulators and metals , author=. J. Chem. Theory Comput. , volume=. 2024 , publisher=
2024
-
[39]
Adsorption and vibrational spectroscopy of
Ye, Hong-Zhou and Berkelbach, Timothy C , journal=. Adsorption and vibrational spectroscopy of. 2024 , publisher=
2024
-
[40]
Ab initio surface chemistry with chemical accuracy , author=. arXiv:2309.14640 , year=
-
[41]
Coupled-cluster singles and doubles for extended systems , author=. J. Chem. Phys. , volume=. 2004 , publisher=. doi:10.1063/1.1637577 , url=
-
[42]
Gaussian-based coupled-cluster theory for the ground-state and band structure of solids , author=. J. Chem. Theory Comput. , volume=. 2017 , publisher=
2017
-
[43]
Coupled cluster theory in materials science , author=. Front. Mater. , volume=. 2019 , publisher=
2019
-
[44]
Applying the Coupled-Cluster Ansatz to Solids and Surfaces in the Thermodynamic Limit , author =. Phys. Rev. X , volume =. 2018 , month =. doi:10.1103/PhysRevX.8.021043 , url =
-
[45]
Tajti, Attila and Szalay, P. J. Chem. Phys. , volume=. 2004 , publisher=
2004
-
[46]
High-accuracy extrapolated ab initio thermochemistry
Bomble, Yannick J and V. High-accuracy extrapolated ab initio thermochemistry. J. Chem. Phys. , volume=. 2006 , publisher=
2006
-
[47]
High-accuracy extrapolated ab initio thermochemistry
Harding, Michael E and V. High-accuracy extrapolated ab initio thermochemistry. J. Chem. Phys. , volume=. 2008 , publisher=
2008
-
[48]
W4 theory for computational thermochemistry: In pursuit of confident sub-
Karton, Amir and Rabinovich, Elena and Martin, Jan ML and Ruscic, Branko , journal=. W4 theory for computational thermochemistry: In pursuit of confident sub-. 2006 , publisher=
2006
-
[49]
Platinum, gold, and silver standards of intermolecular interaction energy calculations , author=. J. Chem. Phys. , volume=. 2019 , publisher=
2019
-
[50]
Understanding discrepancies in noncovalent interaction energies from wavefunction theories for large molecules , author=. Nat. Commun. , volume=. 2025 , publisher=
2025
-
[51]
Interactions between large molecules pose a puzzle for reference quantum mechanical methods , author=. Nat. Commun. , volume=. 2021 , publisher=
2021
-
[52]
Highly accurate electronic structure of metallic solids from coupled-cluster theory with nonperturbative triple excitations , author=. Phys. Rev. Lett. , volume=. 2023 , publisher=
2023
-
[53]
Averting the infrared catastrophe in the gold standard of quantum chemistry , author=. Phys. Rev. Lett. , volume=. 2023 , publisher=
2023
-
[54]
The Journal of Chemical Physics , volume=
Single-reference coupled cluster theory for multi-reference problems , author=. The Journal of Chemical Physics , volume=. 2017 , publisher=
2017
-
[55]
An efficient reformulation of the closed-shell coupled cluster single and double excitation
Scuseria, Gustavo E and Janssen, Curtis L and Schaefer III, Henry F , journal=. An efficient reformulation of the closed-shell coupled cluster single and double excitation. 1988 , publisher=
1988
-
[56]
A direct atomic orbital driven implementation of the coupled cluster singles and doubles
Koch, Henrik and Christiansen, Ove and Kobayashi, Rika and J. A direct atomic orbital driven implementation of the coupled cluster singles and doubles. Chem. Phys. Lett. , volume=. 1994 , publisher=
1994
-
[57]
Linear-Scaling Local Natural Orbital-Based Full Triples Treatment in Coupled-Cluster Theory , author=. J. Chem. Theory Comput. , volume=. 2025 , publisher=
2025
-
[58]
Linear-scaling quadruple excitations in local pair natural orbital coupled-cluster theory , author=. J. Chem. Phys. , volume=. 2025 , publisher=
2025
-
[59]
Local Pair Natural Orbital-Based Coupled-Cluster Theory through Full Quadruples (DLPNO--CCSDTQ) , author=. J. Chem. Theory Comput. , volume=. 2026 , publisher=
2026
-
[60]
Orthogonally-spin-adapted coupled-cluster theory for closed-shell systems including triexcited clusters , author =. Phys. Rev. A , volume =. 1979 , month =. doi:10.1103/PhysRevA.20.1 , url =
-
[61]
Revisitation of nonorthogonal spin adaptation in coupled cluster theory , author=. J. Chem. Theory Comput. , volume=. 2013 , publisher=
2013
-
[62]
Non-orthogonal spin-adaptation of coupled cluster methods: A new implementation of methods including quadruple excitations , author=. J. Chem. Phys. , volume=. 2015 , publisher=
2015
-
[63]
Mathematical Physics in Theoretical Chemistry , publisher=. 2019 , series=. doi:https://doi.org/10.1016/B978-0-12-813651-5.00010-3 , url=
-
[64]
High-performance tensor contraction without transposition , author=. SIAM J. Sci. Comput. , volume=. 2018 , publisher=
2018
-
[65]
Strassen's algorithm for tensor contraction , author=. SIAM J. Sci. Comput. , volume=. 2018 , publisher=
2018
-
[66]
Hillenbrand, Christopher , title=
-
[67]
Matthews, Devin , title=
-
[68]
ACM Trans
Spin Summations: A High-Performance Perspective , author=. ACM Trans. Math. Softw. , volume=. 2019 , publisher=
2019
-
[69]
2018 , publisher=
Sun, Qiming and Berkelbach, Timothy C and Blunt, Nick S and Booth, George H and Guo, Sheng and Li, Zhendong and Liu, Junzi and McClain, James D and Sayfutyarova, Elvira R and Sharma, Sandeep and others , journal=. 2018 , publisher=
2018
-
[70]
Recent developments in the
Sun, Qiming and Zhang, Xing and Banerjee, Samragni and Bao, Peng and Barbry, Marc and Blunt, Nick S and Bogdanov, Nikolay A and Booth, George H and Chen, Jia and Cui, Zhi-Hao and others , journal=. Recent developments in the. 2020 , publisher=
2020
-
[71]
Sun, Qiming and Hermes, Matthew R and Wu, Xiaojie and Zhai, Huanchen and Zhang, Xing and Ahmed, Abdelrahman M and Aucar, Juan Jos. The. arXiv:2603.14155 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[72]
1998 , publisher=
Wales, David J and Hodges, Matthew P , journal=. 1998 , publisher=
1998
-
[73]
Development of local natural orbital arbitrary order coupled cluster methods and assessment through connected quadruples , author=. J. Phys. Chem. A , volume=. 2026 , publisher=
2026
-
[74]
Intermolecular Interactions of Large Systems: Boron Nitrides, Acenes, and Coronenes
Intermolecular Interactions of Large Systems: Boron Nitrides, Acenes, and Coronenes , author=. arXiv preprint arXiv:2602.04723 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[75]
Another angle on benchmarking noncovalent interactions , author=. J. Chem. Theory Comput. , volume=. 2025 , publisher=. doi:10.1021/acs.jctc.4c01512 , url=
-
[76]
When gold is not enough: platinum standard of quantum chemistry with
Lesiuk, Micha. When gold is not enough: platinum standard of quantum chemistry with. J. Chem. Theory Comput. , volume=. 2022 , publisher=
2022
-
[77]
Comprehensive thermochemical benchmark set of realistic closed-shell metal organic reactions , author=. J. Chem. Theory Comput. , volume=. 2018 , publisher=. doi:10.1021/acs.jctc.7b01183 , url=
-
[78]
A simple permutation group approach to spin-free higher-order coupled-cluster methods
A simple permutation group approach to spin-free higher-order coupled-cluster methods , author=. arXiv preprint arXiv:1805.00565 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[79]
and Nguyen, B.N
Goodfellow, A.S. and Nguyen, B.N. , title =. J. Chem. Theory Comput. , volume=. 2026 , doi =
2026
-
[80]
Stochastic tensor contraction for quantum chemistry
Stochastic tensor contraction for quantum chemistry , author=. arXiv:2602.17158 , year=
work page internal anchor Pith review Pith/arXiv arXiv
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.