Recognition: no theorem link
Stochastic tensor contraction for quantum chemistry
Pith reviewed 2026-05-15 21:15 UTC · model grok-4.3
The pith
Stochastic sampling reduces the cost of tensor contractions in coupled cluster calculations to mean-field scaling for high-accuracy energies.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Stochastic tensor contraction performs the tensor operations central to coupled cluster theory by replacing deterministic contractions with stochastic sampling, achieving mean-field computational scaling for total energies more accurate than chemical accuracy while approaching mean-field absolute costs and outperforming local correlation methods in both time and error.
What carries the argument
Stochastic tensor contraction, which approximates high-order tensor contractions via sampling to lower scaling while controlling variance and bias in coupled cluster amplitudes and intermediates.
If this is right
- The scaling of coupled cluster calculations drops to mean-field levels for target accuracies stricter than chemical accuracy.
- Absolute computational cost begins to approach that of mean-field theory.
- Benchmarks show roughly an order of magnitude better computation time and error than state-of-the-art local correlation methods.
- Performance gains hold with less sensitivity to molecular dimensionality or electron delocalization.
- The same primitive can accelerate other quantum chemistry methods that rely on high-order tensor contractions.
Where Pith is reading between the lines
- The stochastic primitive could be combined with existing local approximations to push accuracy-cost trade-offs even further for very large systems.
- Because sampling is embarrassingly parallel, the method may map efficiently onto GPU or distributed architectures.
- Similar stochastic reformulations might apply to tensor contractions in other many-body methods outside chemistry, such as in nuclear physics or condensed matter.
- If variance control generalizes, routine calculations on systems previously limited by cost, like extended molecular chains or clusters, become practical.
Load-bearing premise
The stochastic sampling must converge quickly with controllable variance and negligible bias when applied to the specific tensor structures that appear in coupled cluster amplitudes.
What would settle it
Running the method on a small molecule where the exact deterministic coupled cluster energy is already known and observing either slow variance reduction or a systematic bias exceeding chemical accuracy would falsify the central performance claim.
Figures








read the original abstract
Many computational methods in ab initio quantum chemistry are formulated in terms of high-order tensor contractions, whose cost determines the size of system that can be studied. We introduce stochastic tensor contraction to perform such operations with greatly reduced cost, and present its application to the gold-standard quantum chemistry method, coupled cluster theory with up to perturbative triples. For total energy errors more stringent than chemical accuracy, we reduce the computational scaling to that of mean-field theory, while starting to approach the mean-field absolute cost, thereby challenging the existing cost-to-accuracy landscape. Benchmarks against state-of-the-art local correlation approximations further show that we achieve an order-of-magnitude improvement in both total computation time and error, with significantly reduced sensitivity to system dimensionality and electron delocalization. We conclude that stochastic tensor contraction is a powerful computational primitive to accelerate a wide range of quantum chemistry.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces stochastic tensor contraction as a computational primitive for high-order tensor operations in ab initio quantum chemistry. It applies the method to coupled-cluster theory with perturbative triples and claims that, for total-energy errors stricter than chemical accuracy, the approach reduces computational scaling to mean-field levels while approaching mean-field absolute cost; benchmarks are reported to show an order-of-magnitude improvement over state-of-the-art local correlation methods with reduced sensitivity to dimensionality and delocalization.
Significance. If the stochastic estimators achieve sub-chemical accuracy with sample counts that remain independent of system size, the work would meaningfully expand the reach of high-accuracy correlated methods to larger molecules and alter the existing cost-accuracy frontier. The reported outperformance relative to local approximations and the emphasis on controllable variance constitute potentially important strengths, provided the underlying sampling analysis and scaling data are fully documented.
major comments (2)
- [Abstract and §4 (Benchmarks)] The central claim of mean-field scaling for sub-chemical-accuracy errors rests on the assumption that Monte Carlo variance for CC tensor contractions (amplitudes and intermediates) does not grow with system size. No explicit variance-versus-N plots, sample-complexity bounds, or bias analysis for the specific tensor structures are referenced in the abstract or benchmark summary; this omission is load-bearing and must be addressed with quantitative data.
- [§4 (Benchmarks)] The reported order-of-magnitude improvement over local correlation methods is presented without tabulated error metrics, system sizes, or basis-set details that would allow direct comparison of total wall time versus error (e.g., Table 3 or equivalent). Without these, the cross-method claim cannot be evaluated for generality.
minor comments (2)
- [§2] Notation for the stochastic estimator (e.g., definition of the contraction primitive and its variance) should be introduced with a clear equation early in the methods section to aid readability.
- [Figures 2-4] Figure captions for scaling plots should explicitly state the number of samples used and the target error threshold to make the mean-field scaling claim immediately verifiable.
Simulated Author's Rebuttal
We thank the referee for the careful and constructive review. We address each major comment below and have revised the manuscript to incorporate additional quantitative data on variance scaling and benchmark details.
read point-by-point responses
-
Referee: [Abstract and §4 (Benchmarks)] The central claim of mean-field scaling for sub-chemical-accuracy errors rests on the assumption that Monte Carlo variance for CC tensor contractions (amplitudes and intermediates) does not grow with system size. No explicit variance-versus-N plots, sample-complexity bounds, or bias analysis for the specific tensor structures are referenced in the abstract or benchmark summary; this omission is load-bearing and must be addressed with quantitative data.
Authors: We agree that explicit demonstration of the variance scaling is essential to support the mean-field claim. In the revised manuscript we have added new figures in §4 that plot Monte Carlo variance versus system size N for the key CC tensor contractions (T2 amplitudes, T3 intermediates, and energy contributions). These data confirm that variance remains controlled such that sub-chemical accuracy is maintained with sample counts that preserve overall mean-field scaling. A concise bias analysis and sample-complexity discussion have also been inserted in the Methods section, and the abstract now references these supporting results. revision: yes
-
Referee: [§4 (Benchmarks)] The reported order-of-magnitude improvement over local correlation methods is presented without tabulated error metrics, system sizes, or basis-set details that would allow direct comparison of total wall time versus error (e.g., Table 3 or equivalent). Without these, the cross-method claim cannot be evaluated for generality.
Authors: We accept that the comparison requires more granular tabulated information. The revised §4 now includes an expanded Table 3 (plus supplementary tables) that list, for each benchmark system, the total energy error, system size, basis set, wall-clock time, and number of samples for both stochastic tensor contraction and the local correlation methods. Additional text discusses the observed order-of-magnitude gains in time and error across the tested range of dimensionality and delocalization. revision: yes
Circularity Check
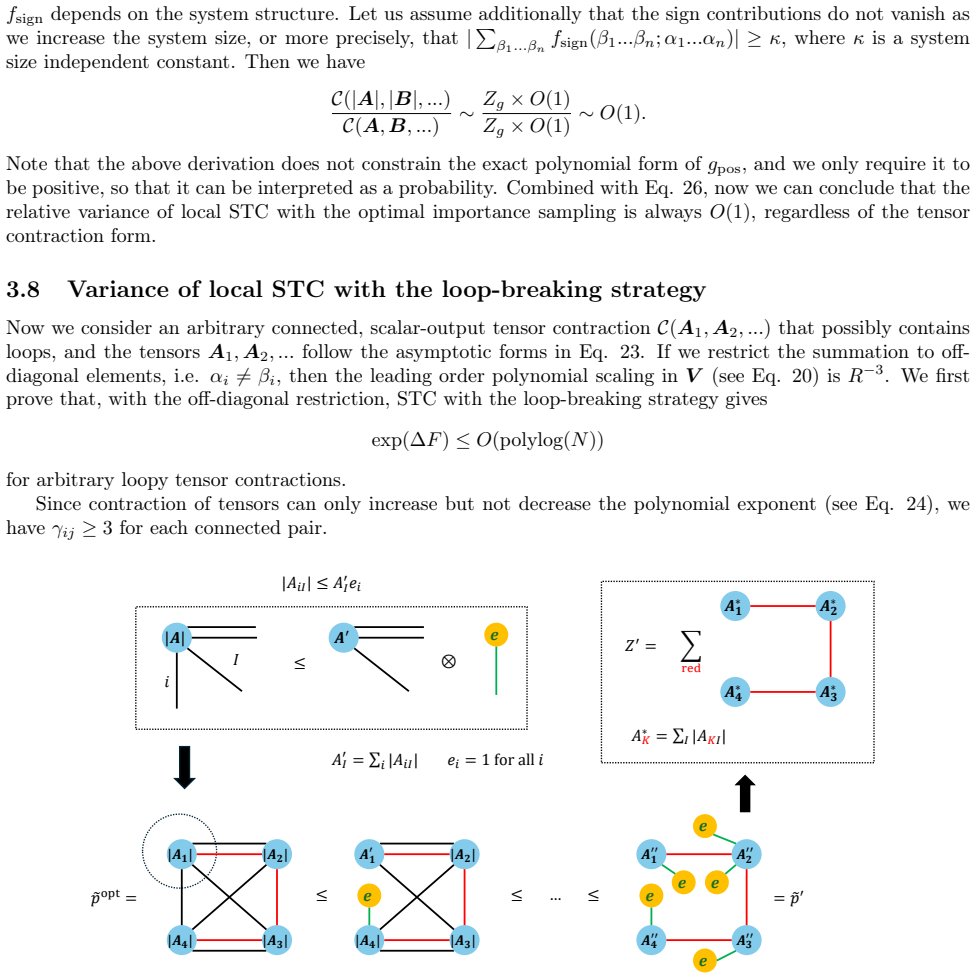
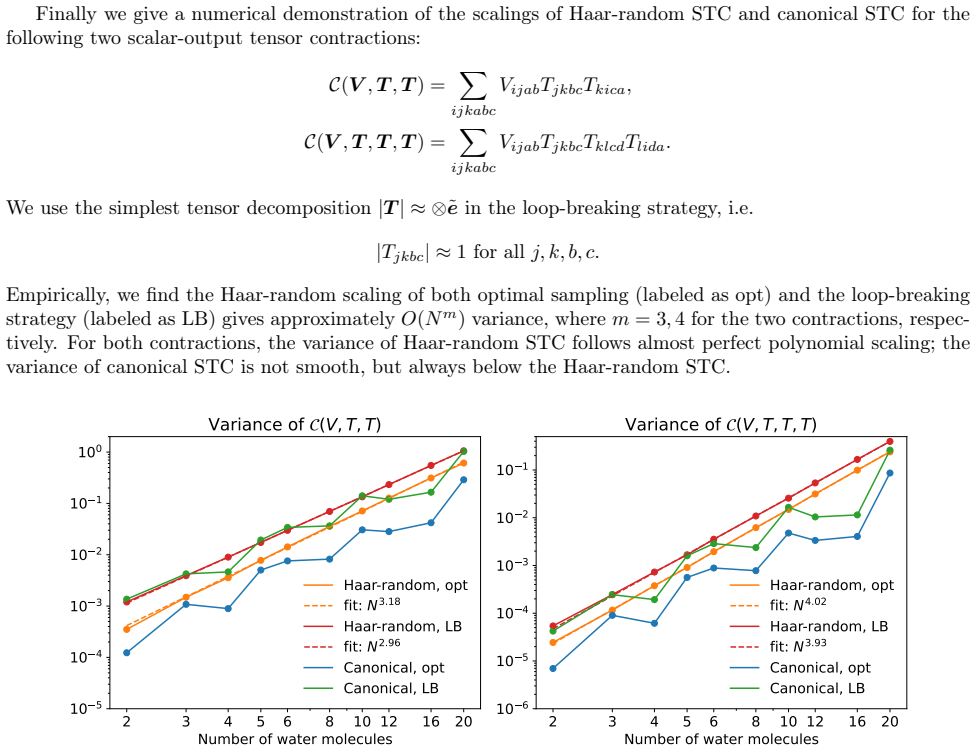
No circularity; stochastic contraction scaling derived from independent Monte Carlo variance bounds
full rationale
The paper introduces stochastic tensor contraction as a new primitive whose cost scaling follows from the number of samples needed to control variance in tensor contractions for CC amplitudes and intermediates. No equations in the abstract or described derivation reduce the claimed mean-field scaling to a fitted parameter, self-defined quantity, or prior self-citation chain. The method is benchmarked against external local approximations without the central result being forced by those inputs. The variance control assumption is presented as an independent property of the stochastic estimator rather than tautological with the target accuracy.
Axiom & Free-Parameter Ledger
Forward citations
Cited by 1 Pith paper
-
Stochastic Loop Corrections to Belief Propagation for Tensor Network Contraction
A stochastic MCMC sampling method with umbrella sampling provides unbiased loop corrections to belief propagation for exact factorization-based tensor network contraction on loopy graphs with symmetric potentials.
Reference graph
Works this paper leans on
-
[1]
(Cambridge university press), (2009)
I Shavitt, RJ Bartlett, Many-body methods in chemistry and physics: MBPT and coupled-cluster theory . (Cambridge university press), (2009)
work page 2009
-
[2]
T Helgaker, P Jorgensen, J Olsen, Molecular electronic-structure theory . (John Wiley & Sons), (2013)
work page 2013
-
[3]
RJ Bartlett, M Musiał, Coupled-cluster theory in quantum chemistry . Rev. Mod. Phys. 79, 291–352 (2007)
work page 2007
-
[4]
a general technique for determining electron correlation energies
JA Pople, M Head-Gordon, K Raghavachari, Quadratic configuration interaction. a general technique for determining electron correlation energies. The J. chemical physics 87, 5968–5975 (1987)
work page 1987
-
[5]
A Dreuw, M Head-Gordon, Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. reviews 105, 4009–4037 (2005)
work page 2005
-
[6]
GP Chen, VK Voora, MM Agee, SG Balasubramani, F Furche, Random-phase approximation methods. Annu. Rev. Phys. Chem . 68, 421–445 (2017)
work page 2017
-
[7]
S Sharma, AA Holmes, G Jeanmairet, A Alavi, CJ Umrigar, Semistochastic heat-bath configuration interaction method: Selected configuration interaction with semistochastic perturbation theory . J. chemical theory computation 13, 1595–1604 (2017)
work page 2017
-
[8]
The journal physical chemistry letters 12, 12084–12097 (2021)
J Shee, M Loipersberger, A Rettig, J Lee, M Head-Gordon, Regularized second-order møller–plesset theory: A more accurate alternative to conventional mp2 for noncovalent interactions and transition metal thermochemistry for the same computational cost. The journal physical chemistry letters 12, 12084–12097 (2021)
work page 2021
-
[9]
A Dreuw, M Wormit, The algebraic diagrammatic construction scheme for the polarization propagator for the calculation of excited states. Wiley Interdiscip. Rev. Comput. Mol. Sci . 5, 82–95 (2015)
work page 2015
-
[10]
MJ van Setten, F Weigend, F Evers, The gw-method for quantum chemistry applications: Theory and implementation. J. chemical theory computation 9, 232–246 (2013). 11. K Raghavachari, GW T rucks, JA Pople, M Head-Gordon, A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett . 157, 479–483 (1989)
work page 2013
-
[11]
energies and analytical gradients
JD Watts, J Gauss, RJ Bartlett, Coupled-cluster methods with noniterative triple excitations for restricted open-shell hartree–fock and other general single determinant reference functions. energies and analytical gradients. The J. chemical physics 98, 8718–8733 (1993). 13. M Urban, J Noga, SJ Cole, RJ Bartlett, T owards a full ccsdt model for electron co...
work page 1993
-
[12]
J Y ang, et al., Ab initio determination of the crystalline benzene lattice energy to sub-kilojoule/mole accuracy . Science 345, 640–643 (2014)
work page 2014
-
[13]
BX Shi, et al., An accurate and efficient framework for modelling the surface chemistry of ionic materials. Nat. chemistry 17, 1688–1695 (2025)
work page 2025
-
[14]
GE Scuseria, PY Ayala, Linear scaling coupled cluster and perturbation theories in the atomic orbital basis. The J. chemical physics 111, 8330–8343 (1999)
work page 1999
-
[15]
M Sch ¨utz, HJ Werner, Low-order scaling local electron correlation methods. iv. linear scaling local coupled-cluster (lccsd). The J. Chem. Phys . 114, 661–681 (2001)
work page 2001
-
[16]
J Y ang, GK Chan, FR Manby , M Sch ¨utz, HJ Werner, The orbital-specific-virtual local coupled cluster singles and doubles method. The J. Chem. Phys . 136 (2012)
work page 2012
-
[17]
M Sch ¨utz, J Y ang, GK Chan, FR Manby , HJ Werner, The orbital-specific virtual local triples correction: Osv-l (t). The J. Chem. Phys . 138 (2013)
work page 2013
-
[18]
F Neese, A Hansen, DG Liakos, Efficient and accurate approximations to the local coupled cluster singles doubles method using a truncated pair natural orbital basis. The J. chemical physics 131 (2009)
work page 2009
-
[19]
F Neese, F Wennmohs, A Hansen, Efficient and accurate local approximations to coupled-electron pair approaches: An attempt to revive the pair natural orbital method. The J. chemical physics 130 (2009)
work page 2009
-
[20]
C Riplinger, P Pinski, U Becker, EF Valeev, F Neese, Sparse maps—a systematic infrastructure for reduced-scaling electronic structure methods. ii. linear scaling domain based pair natural orbital coupled cluster theory . The J. chemical physics 144 (2016)
work page 2016
-
[21]
DG Liakos, Y Guo, F Neese, Comprehensive benchmark results for the domain based local pair natural orbital coupled cluster method (dlpno-ccsd (t)) for closed-and open-shell systems. The J. Phys. Chem. A 124, 90–100 (2019)
work page 2019
-
[22]
PR Nagy , G Samu, M K ´allay , Optimization of the linear-scaling local natural orbital ccsd (t) method: Improved algorithm and benchmark applications. J. Chem. Theory Comput . 14, 4193–4215 (2018)
work page 2018
-
[23]
PR Nagy , M K´allay , Approaching the basis set limit of ccsd (t) energies for large molecules with local natural orbital coupled-cluster methods. J. Chem. Theory Comput . 15, 5275–5298 (2019)
work page 2019
-
[24]
RM Parrish, CD Sherrill, EG Hohenstein, SI Kokkila, TJ Mart ´ınez, Communication: Acceleration of coupled cluster singles and doubles via orbital-weighted least-squares tensor hypercontraction. The J. Chem. Phys . 140 (2014)
work page 2014
-
[25]
A Jiang, JM T urney , HF Schaefer III, T ensor hypercontraction form of the perturbative triples energy in coupled-cluster theory . J. chemical theory computation 19, 1476–1486 (2023)
work page 2023
-
[26]
S Saebo, P Pulay , Local treatment of electron correlation. Annu. Rev. Phys. Chem . 44, 213–236 (1993)
work page 1993
-
[27]
J Lee, HQ Pham, DR Reichman, T wenty years of auxiliary-field quantum monte carlo in quantum chemistry: An overview and assessment on main group chemistry and bond-breaking. J. Chem. Theory Comput . 18, 7024–7042 (2022). 30. M Motta, S Zhang, Ab initio computations of molecular systems by the auxiliary-field quantum monte carlo method. Wiley Interdiscip. Re...
work page 2022
-
[28]
BM Austin, DY Zubarev, WA Lester Jr, Quantum monte carlo and related approaches. Chem. reviews 112, 263–288 (2012)
work page 2012
-
[29]
J Hermann, Z Sch ¨atzle, F No ´e, Deep-neural-network solution of the electronic schr ¨odinger equation. Nat. Chem. 12, 891–897 (2020)
work page 2020
-
[30]
AJW Thom, A Alavi, Stochastic perturbation theory: A low-scaling approach to correlated electronic energies. Phys. Rev. Lett. 99, 143001 (2007)
work page 2007
-
[31]
AJW Thom, Stochastic coupled cluster theory . Phys. Rev. Lett. 105, 263004 (2010)
work page 2010
-
[32]
D Neuhauser, E Rabani, R Baer, Expeditious stochastic approach for mp2 energies in large electronic systems. J. Chem. theory Comput . 9, 24–27 (2013)
work page 2013
-
[33]
R Baer, D Neuhauser, E Rabani, Stochastic vector techniques in ground-state electronic structure. Annu. Rev. Phys. Chem . 73, 255–272 (2022)
work page 2022
-
[34]
SY Willow, KS Kim, S Hirata, Stochastic evaluation of second-order many-body perturbation energies. The J. chemical physics 137 (2012)
work page 2012
-
[35]
Y Damour, A Gallo, A Scemama, Stochastically accelerated perturbative triples correction in coupled cluster calculations. The J. Chem. Phys . 161 (2024)
work page 2024
-
[36]
IEEE T ransactions on software engineering 17, 972 (1991)
MD Vose, A linear algorithm for generating random numbers with a given distribution. IEEE T ransactions on software engineering 17, 972 (1991)
work page 1991
-
[37]
JS Y edidia, WT Freeman, Y Weiss, , et al., Understanding belief propagation and its generalizations. Explor. artificial intelligence new millennium 8, 0018–9448 (2003)
work page 2003
-
[38]
Loopy Belief Propagation for Approximate Inference: An Empirical Study
K Murphy , Y Weiss, MI Jordan, Loopy belief propagation for approximate inference: An empirical study . arXiv preprint arXiv:1301.6725 (2013)
work page internal anchor Pith review Pith/arXiv arXiv 2013
-
[39]
A Paszke, et al., Pytorch: An imperative style, high-performance deep learning library . Adv. neural information processing systems 32 (2019)
work page 2019
-
[40]
Q Sun, et al., Pyscf: the python-based simulations of chemistry framework. Wiley Interdiscip. Rev. Comput. Mol. Sci . 8, e1340 (2018)
work page 2018
-
[41]
Q Sun, et al., Recent developments in the pyscf program package. The J. chemical physics 153 (2020)
work page 2020
-
[42]
F Neese, F Wennmohs, U Becker, C Riplinger, The orca quantum chemistry program package. The J. chemical physics 152 (2020)
work page 2020
-
[43]
WJ Hehre, R Ditchfield, JA Pople, Self—consistent molecular orbital methods. xii. further extensions of gaussian—type basis sets for use in molecular orbital studies of organic molecules. The J. Chem. Phys . 56, 2257–2261 (1972)
work page 1972
-
[44]
TH Dunning Jr, Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen. The J. chemical physics 90, 1007–1023 (1989). 48. J VandeVondele, J Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. The J. chemical physics 127 (2007)
work page 1989
-
[45]
systematic basis sets and wave functions
RA Kendall, TH Dunning Jr, RJ Harrison, Electron affinities of the first-row atoms revisited. systematic basis sets and wave functions. The J. chemical physics 96, 6796–6806 (1992)
work page 1992
-
[46]
D Datta, S Kossmann, F Neese, Analytic energy derivatives for the calculation of the first-order molecular properties using the domain-based local pair-natural orbital coupled-cluster theory . The J. Chem. Phys . 145 (2016)
work page 2016
-
[47]
PJ Stephens, FJ Devlin, CF Chabalowski, MJ Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. The J. physical chemistry 98, 11623–11627 (1994)
work page 1994
-
[48]
N Mardirossian, M Head-Gordon, ω b97m-v: A combinatorially optimized, range-separated hybrid, meta-gga density functional with vv10 nonlocal correlation. The J. Chem. Phys . 144, 214110 (2016). 10 of 10 www.pnas.org/cgi/doi/10.1073/pnas.XXXXXXXXXX pnas.org Support information for Stochastic tensor contraction for quantum chemistry Jiace Sun 1,2, Garnet Ki...
-
[49]
As proved in Sec. 3.6, extra poly (N ) variance scalings only possibly appear when some tensors have γ = 1, 2 decay. In CCD, only the three terms in the second line of Eq. 34 have γ = 1 . However, all of them lead to a next-iteration energy diagram exactly the same as the left one shown in Fig. 3, thus in fact they do not contribute extra poly (N ) factor...
-
[50]
Thus for ˜p′ we also have exp(∆ F ) ∼ O(N )
PP abc simply contributes a multiplication factor of 36 = 6 × 6. Thus for ˜p′ we also have exp(∆ F ) ∼ O(N ). Thus the final deterministic cost, and sampling variance have the same scaling with the shown representative tensor contraction term in the main text. We have already shown in the main text that ˜p′ 1 (and ˜p′ 2 similarly) supports efficient sampling...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.