Differentiable inverse design of short-range order in high-entropy alloys: from target sro to target property
Pith reviewed 2026-07-03 09:34 UTC · model grok-4.3
The pith
Gradient optimization designs SRO in alloys to hit target stiffness
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By making atom occupancy continuous and applying gradient descent with an extended physics-based correction term, the method generates thermodynamically realistic alloy structures that match target short-range order and produce specified mechanical properties, forming a closed pipeline from target property back to atomic arrangement.
What carries the argument
Continuous atom-occupancy representation optimized by gradient descent, paired with an extended physics-based correction term for thermodynamic realism and a neural-network property predictor.
If this is right
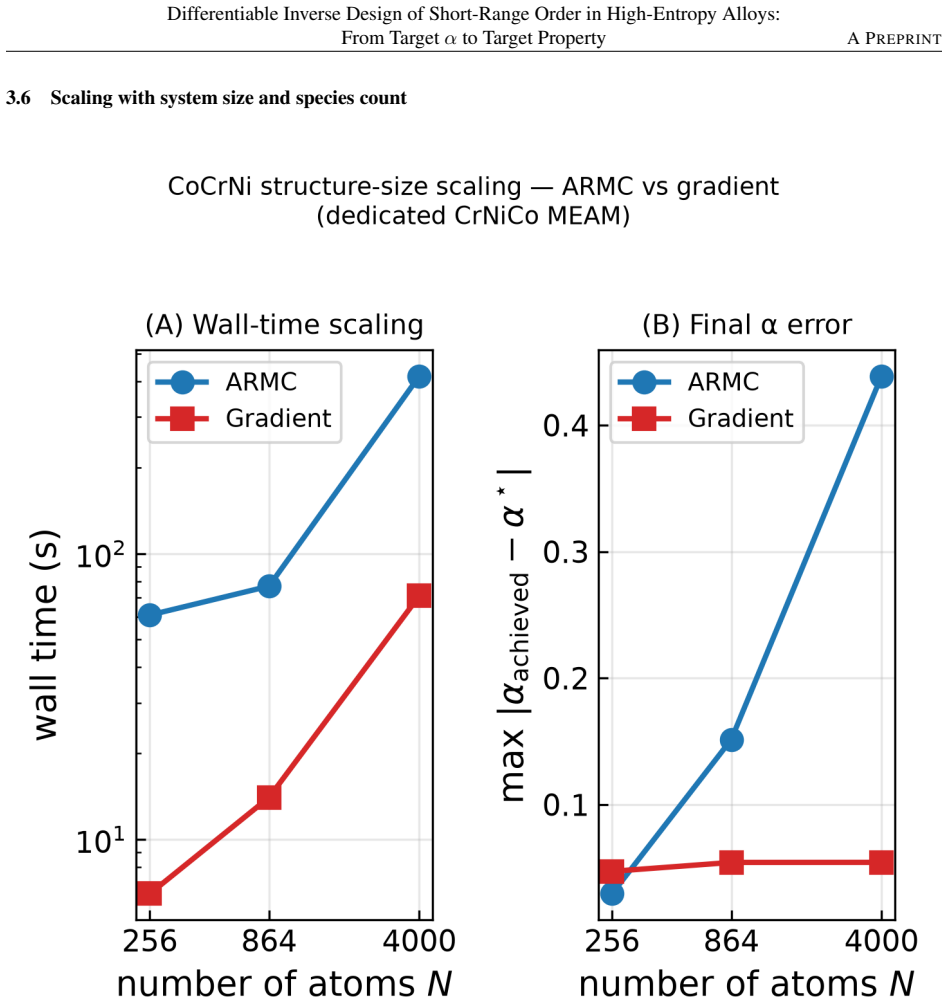
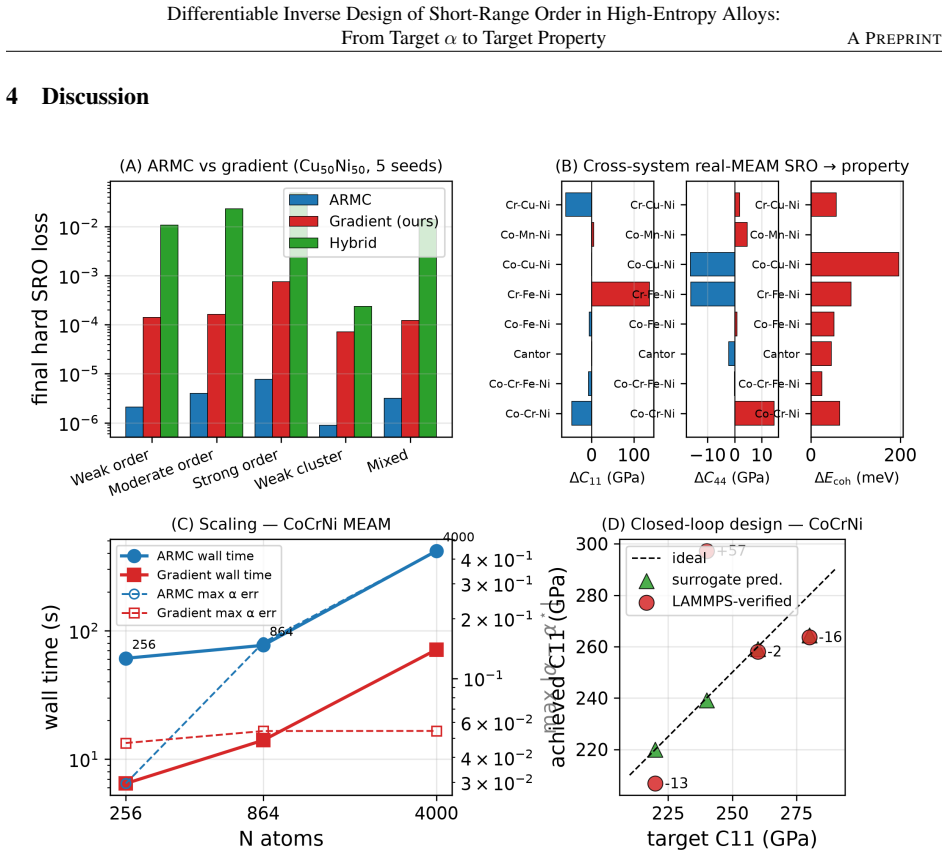
- The builder matches random-swap accuracy on small systems but runs six times faster and eight times more accurately on 4000-atom systems.
- Simulation cells need at least 864 atoms to capture the correct direction and magnitude of SRO-driven stiffness changes.
- The approach scales smoothly to alloys with many elements without added bookkeeping.
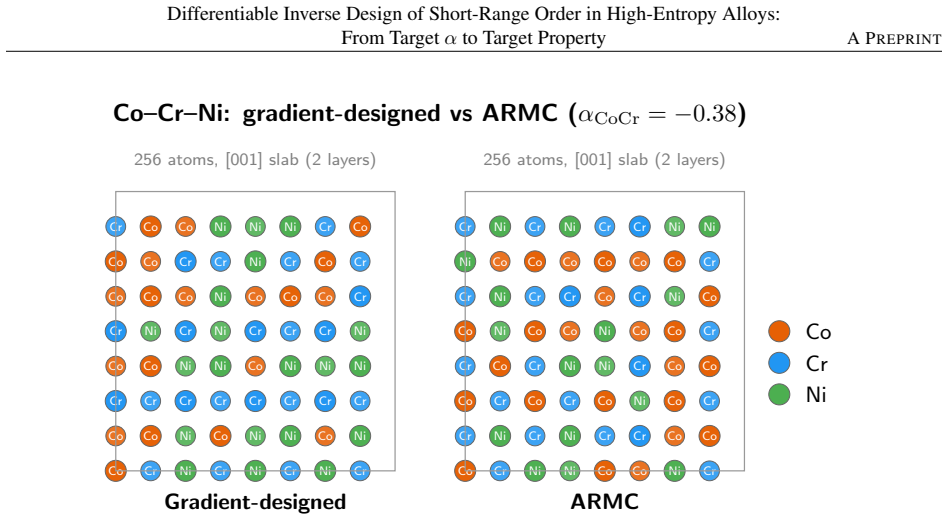
- It reproduced three of four target stiffness values within 6% when checked against real simulations for a cobalt-chromium-nickel alloy.
Where Pith is reading between the lines
- The same continuous-occupancy gradient framework could be retrained on other properties such as yield strength or thermal conductivity to expand the design targets.
- Literature results based on 108-atom cells may systematically under- or over-estimate SRO effects on properties.
- Releasing the method as open-source Python code allows direct testing on new alloy families and property combinations.
Load-bearing premise
That extending the physics-based correction term from two-element to many-element alloys sufficiently enforces thermodynamic realism in the optimized structures rather than merely matching numerical SRO targets.
What would settle it
Running independent molecular dynamics simulations on the designed structures for additional alloys beyond the nine tested and checking whether the resulting stiffness values match the pipeline predictions within 6%.
Figures









read the original abstract
Short-range order (SRO) governs the mechanical response of multi-principal-element alloys, but designing an alloy for a target property usually means solving two disconnected problems: building a structure matching a desired SRO pattern, then separately checking its property, with no shared optimization. This work replaces the standard random-swap search (reverse Monte Carlo) with a gradient-based approach: atom occupancy is treated as continuous rather than fixed, so the whole process can be tuned using gradient descent, the same method used to train neural networks. This builder matches random-swap accuracy on small systems, but is six times faster and eight times more accurate on large 4000-atom systems, and scales smoothly to alloys with many elements without extra bookkeeping. A physics-based correction term, adapted from prior two-element work and extended here to many elements, keeps designed structures thermodynamically realistic rather than just numerically matching the target SRO pattern. A small neural network then predicts mechanical properties directly from composition and SRO statistics, closing the loop from target property back to structure. Tested on nine face-centered-cubic and body-centered-cubic alloys, the pipeline captured SRO-driven stiffness changes from -20% to +57%, and cell-size checks showed at least 864 atoms are needed to get the direction and size of these changes right, since the commonly used 108-atom cells can mislead. Against real simulations for a cobalt-chromium-nickel alloy, the method matched three of four target stiffness values within 6%. The method is released as an open-source Python package, anisro, offering a practical route to gradient-based, property-driven alloy design.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims a gradient-based inverse-design pipeline for short-range order (SRO) in high-entropy alloys that treats atom occupancy as continuous, optimizes via gradient descent, employs a neural-network property predictor, and uses an extended physics-based correction term (adapted from two-element prior work) to enforce thermodynamic realism. On nine FCC and BCC alloys the method is reported to recover SRO-driven stiffness changes of -20% to +57%, requires cells of at least 864 atoms, and matches three of four target stiffness values within 6% for CoCrNi; the code is released as the open-source package anisro.
Significance. If the thermodynamic-realism claim holds, the work supplies a scalable, differentiable route from target property to SRO structure that is substantially faster and more accurate than reverse Monte Carlo on large cells and removes the need for disconnected forward and inverse steps. The open-source release is a concrete strength that supports reproducibility.
major comments (2)
- [Abstract] Abstract (description of the correction term): the claim that the physics-based correction term, when extended to many elements, keeps structures 'thermodynamically realistic rather than just numerically matching the target SRO pattern' is load-bearing for the central assertion that the pipeline yields property-relevant SRO. No explicit multi-element energy functional, derivation, or validation against independent Monte Carlo pair-correlation thermodynamics or formation energies is supplied; without this the extension risks reducing to a soft constraint on Warren-Cowley parameters.
- [Abstract] Abstract (performance claims): the statements of 'six times faster and eight times more accurate on large 4000-atom systems' and 'at least 864 atoms are needed' are presented without error bars, dataset descriptions, or cross-validation details. These metrics are central to the superiority claim over random-swap search and must be supported by explicit methods and statistics.
minor comments (1)
- The abstract reports concrete numerical matches (6% on CoCrNi) but supplies no description of the neural-network architecture, training set size, or loss function; these details belong in the main text for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments on our manuscript. We address each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract (description of the correction term): the claim that the physics-based correction term, when extended to many elements, keeps structures 'thermodynamically realistic rather than just numerically matching the target SRO pattern' is load-bearing for the central assertion that the pipeline yields property-relevant SRO. No explicit multi-element energy functional, derivation, or validation against independent Monte Carlo pair-correlation thermodynamics or formation energies is supplied; without this the extension risks reducing to a soft constraint on Warren-Cowley parameters.
Authors: We agree that the abstract claim requires explicit supporting material. The correction term is an extension of the two-element formulation, but the manuscript does not currently provide the multi-element energy functional, its derivation, or direct validation against independent Monte Carlo thermodynamics. In the revised manuscript we will add a dedicated subsection in Methods with the explicit functional form and a new figure comparing pair correlations and formation energies of the optimized structures to independent Monte Carlo results on the same systems. revision: yes
-
Referee: [Abstract] Abstract (performance claims): the statements of 'six times faster and eight times more accurate on large 4000-atom systems' and 'at least 864 atoms are needed' are presented without error bars, dataset descriptions, or cross-validation details. These metrics are central to the superiority claim over random-swap search and must be supported by explicit methods and statistics.
Authors: We acknowledge that the performance statements in the abstract are presented without the requested statistical support. In the revised manuscript we will expand the Results section to report error bars from repeated runs, describe the full dataset (alloys and cell sizes tested), and detail the cross-validation procedure used to quantify the speedup and accuracy gains on 4000-atom systems as well as the cell-size convergence analysis. revision: yes
Circularity Check
No significant circularity; derivation remains self-contained
full rationale
The pipeline consists of a gradient-based optimizer on continuous atom occupancies, a correction term extended from prior two-element work (explicitly stated as adapted and extended in the present manuscript), and a separately trained neural network for property prediction from composition and SRO statistics. No equation or step reduces by construction to a fitted input renamed as output, nor does any load-bearing premise collapse to an unverified self-citation chain. The central loop from target property to structure is externally falsifiable via the reported comparisons to Monte Carlo and experimental stiffness values, satisfying the criteria for independent content.
Axiom & Free-Parameter Ledger
free parameters (2)
- neural-network weights
- correction-term coefficients
axioms (2)
- domain assumption Continuous relaxation of atom occupancy remains valid for producing thermodynamically realistic structures when combined with the physics correction.
- domain assumption The neural network provides a sufficiently accurate forward map from SRO to stiffness for the inverse-design loop to be useful.
Reference graph
Works this paper leans on
-
[1]
Allen, Tatiana Lopez-Guevara, Kimberly Stachenfeld, Alvaro Sanchez-Gonzalez, Peter Battaglia, Jessica Hamrick, and Tobias Pfaff
Kelsey R. Allen, Tatiana Lopez-Guevara, Kimberly Stachenfeld, Alvaro Sanchez-Gonzalez, Peter Battaglia, Jessica Hamrick, and Tobias Pfaff. Physical design using differentiable learned simulators, 2022
2022
-
[2]
Mattias ngqvist, William A. Mu \ n oz, J. Magnus Rahm, Erik Fransson, C \'e line Durniak, Piotr Rozyczko, Thomas H. Rod, and Paul Erhart. ICET -- a python library for constructing and sampling alloy cluster expansions. Advanced Theory and Simulations, 2 0 (7): 0 1900015, 2019. doi:10.1002/adts.201900015
-
[3]
Edwin Antillon, C. Woodward, S. I. Rao, B. Akdim, and T. A. Parthasarathy. Chemical short range order strengthening in a model FCC high entropy alloy. Acta Materialia, 190: 0 29--42, 2020. doi:10.1016/j.actamat.2020.02.041
-
[4]
Kov \'a cs, Gregor N
Ilyes Batatia, D \'a vid P. Kov \'a cs, Gregor N. C. Simm, Christoph Ortner, and G \'a bor Cs \'a nyi. MACE : Higher order equivariant message passing neural networks for fast and accurate force fields. In Advances in Neural Information Processing Systems (NeurIPS), volume 35, 2022
2022
-
[5]
Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P. Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E. Smidt, and Boris Kozinsky. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nature Communications, 13: 0 2453, 2022. doi:10.1038/s41467-022-29939-5
-
[6]
Structural relaxation made simple
Erik Bitzek, Pekka Koskinen, Franz G \"a hler, Michael Moseler, and Peter Gumbsch. Structural relaxation made simple. Physical Review Letters, 97: 0 170201, 2006. doi:10.1103/PhysRevLett.97.170201
-
[7]
B. Cantor, I. T. H. Chang, P. Knight, and A. J. B. Vincent. Microstructural development in equiatomic multicomponent alloys. Materials Science and Engineering: A, 375--377: 0 213--218, 2004. doi:10.1016/j.msea.2003.10.257
-
[8]
Mechanical properties of high-entropy alloys with compositional disorder at small scales
Penghui Cao. Mechanical properties of high-entropy alloys with compositional disorder at small scales. npj Computational Materials, 7: 0 166, 2021. doi:10.1038/s41524-021-00647-y
-
[9]
Direct observation of chemical short-range order in a medium-entropy alloy
Xuefei Chen, Qin Wang, Zhiying Cheng, Mingli Zhu, Hao Zhou, Ping Jiang, Lingling Zhou, Qiqige Xue, Fuping Yuan, Jing Zhu, Xiaolei Wu, and En Ma. Direct observation of chemical short-range order in a medium-entropy alloy. Nature, 592: 0 712--716, 2021. doi:10.1038/s41586-021-03428-z
-
[10]
Structure motif of chemical short-range order in a medium-entropy alloy
Xuefei Chen, Fuping Yuan, Hao Zhou, and Xiaolei Wu. Structure motif of chemical short-range order in a medium-entropy alloy. Materials Research Letters, 10: 0 149--155, 2022. doi:10.1080/21663831.2022.2029607
-
[11]
Won-Mi Choi, Yong Hee Jo, Seok Su Sohn, Sunghak Lee, and Byeong-Joo Lee. Understanding the physical metallurgy of the CoCrFeMnNi high-entropy alloy: an atomistic simulation study. npj Computational Materials, 4: 0 1, 2018. doi:10.1038/s41524-018-0060-9
-
[12]
J. M. Cowley. An approximate theory of order in alloys. Physical Review, 77: 0 669--675, 1950. doi:10.1103/PhysRev.77.669
-
[13]
Bowen Deng, Peichen Zhong, KyuJung Jun, Janosh Riebesell, Kevin Han, Christopher J. Bartel, and Gerbrand Ceder. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nature Machine Intelligence, 5: 0 1031--1041, 2023. doi:10.1038/s42256-023-00716-3
-
[14]
Jun Ding, Qin Yu, Mark Asta, and Robert O. Ritchie. Tunable stacking fault energies by tailoring local chemical order in CrCoNi medium-entropy alloys. Proceedings of the National Academy of Sciences, 115: 0 8919--8924, 2018. doi:10.1073/pnas.1808660115
-
[15]
Torchmd: A deep learning framework for molecular simulations
Stefan Doerr, Maciej Majewski, Adrià Pérez, Andreas Krämer, Cecilia Clementi, Frank Noe, Toni Giorgino, and Gianni De Fabritiis. Torchmd: A deep learning framework for molecular simulations. Journal of Chemical Theory and Computation, 17: 0 2355--2363, 2021. doi:10.1021/acs.jctc.0c01343
-
[16]
Ant \' o nio Fern \'a ndez-Caballero, Marcel H. F. Sluiter, Jakub S. Wrobel, Duc Nguyen-Manh, Sergei L. Dudarev, and Paul M. Mummery. Short-range order strengthening in BCC transition-metal alloys. Physical Review Materials, 4: 0 023403, 2020. doi:10.1103/PhysRevMaterials.4.023403
-
[17]
E. P. George, D. Raabe, and R. O. Ritchie. High-entropy alloys. Nature Reviews Materials, 4: 0 515--534, 2019. doi:10.1038/s41578-019-0121-4
-
[18]
Carl P. Goodrich, Ella M. King, Samuel S. Schoenholz, Ekin D. Cubuk, and Michael P. Brenner. Designing self-assembling kinetics with differentiable statistical physics models. Proceedings of the National Academy of Sciences, 118 0 (10): 0 e2024083118, 2021. doi:10.1073/pnas.2024083118
-
[19]
Capturing short-range order in high-entropy alloys with machine learning potentials
Ying Han et al. Capturing short-range order in high-entropy alloys with machine learning potentials. npj Computational Materials, 11: 0 22, 2025. doi:10.1038/s41524-025-01722-2
-
[20]
Chao Jiang and Blas P. Uberuaga. Efficient ab initio modeling of random multicomponent alloys. Physical Review Letters, 116: 0 105501, 2016. doi:10.1103/PhysRevLett.116.105501
-
[21]
Adam: A Method for Stochastic Optimization
Diederik P. Kingma and Jimmy Ba. Adam: A method for stochastic optimization. In International Conference on Learning Representations (ICLR), 2015. URL https://arxiv.org/abs/1412.6980
work page internal anchor Pith review Pith/arXiv arXiv 2015
-
[22]
Wan-Sik Ko, Blazej Grabowski, and J \"o rg Neugebauer. Development and application of a Ni-Ti interatomic potential with high predictive accuracy of the martensitic phase transition. Physical Review B, 92: 0 134107, 2021. doi:10.1103/PhysRevB.92.134107
-
[23]
Guillaume Laplanche, Pascal Gadaud, Moritz H \"a rtelt, and Easo P. George. Single crystal elastic constants of equiatomic CrCoNi medium-entropy alloy. Materials Science and Engineering: A, 823: 0 141762, 2021. doi:10.1016/j.msea.2021.141762
-
[24]
Byeong-Joo Lee and Michael I. Baskes. Second nearest-neighbor modified embedded-atom-method potential. Physical Review B, 62: 0 8564--8567, 2000. doi:10.1103/PhysRevB.62.8564
-
[25]
Q.-J. Li, H. Sheng, and E. Ma. Strengthening in multi-principal element alloys with local-chemical-order roughened dislocation pathways. Nature Communications, 10: 0 3563, 2019. doi:10.1038/s41467-019-11464-7
-
[26]
Machine learning modeling of high entropy alloy: the role of short-range order
Xianglin Liu, Jiaxin Zhang, and Yang Wang. Machine learning modeling of high entropy alloy: the role of short-range order. arXiv preprint arXiv:1906.10889, 2019
-
[27]
N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller. Equation of state calculations by fast computing machines. The Journal of Chemical Physics, 21: 0 1087--1092, 1953. doi:10.1063/1.1699114
-
[28]
D. B. Miracle and O. N. Senkov. A critical review of high entropy alloys and related concepts. Acta Materialia, 122: 0 448--511, 2017. doi:10.1016/j.actamat.2016.08.081
-
[29]
Owen, Mordechai Kornbluth, and Boris Kozinsky
Albert Musaelian, Simon Batzner, Anders Johansson, Lixin Sun, Cameron J. Owen, Mordechai Kornbluth, and Boris Kozinsky. Learning local equivariant representations for large-scale atomistic dynamics. Nature Communications, 14: 0 579, 2023. doi:10.1038/s41467-023-36329-y
-
[30]
PyTorch: An Imperative Style, High-Performance Deep Learning Library
A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, et al. PyTorch : An imperative style, high-performance deep learning library. In Advances in Neural Information Processing Systems, volume 32, 2019. URL https://arxiv.org/abs/1912.01703
work page internal anchor Pith review Pith/arXiv arXiv 2019
-
[32]
Youfu Rao and William A. Curtin. Analytical models of short-range order in FCC and BCC alloys. Acta Materialia, 226: 0 117621, 2022 b . doi:10.1016/j.actamat.2022.117621
-
[33]
J. M. Sanchez, F. Ducastelle, and D. Gratias. Generalized cluster description of multicomponent systems. Physica A, 128 0 (1--2): 0 334--350, 1984. doi:10.1016/0378-4371(84)90096-7
-
[34]
O. Schneeweiss, M. Fri\' a k, M. Pola s kov\' a , M. Sob, D. Vojt e ch, F. Zelenka, and J. Ve s t\' a l. Magnetic properties of the CrMnFeCoNi high-entropy alloy. Physical Review B, 96: 0 014437, 2017. doi:10.1103/PhysRevB.96.014437
-
[35]
Schoenholz and Ekin D
Samuel S. Schoenholz and Ekin D. Cubuk. JAX MD : A framework for differentiable physics. In Advances in Neural Information Processing Systems (NeurIPS), volume 33, 2020
2020
-
[36]
Chemical-motif characterization of short-range order with E(3) -equivariant graph neural networks
Killian Sheriff, Yifan Cao, and Rodrigo Freitas. Chemical-motif characterization of short-range order with E(3) -equivariant graph neural networks. npj Computational Materials, 10: 0 215, 2024 a . doi:10.1038/s41524-024-01393-5
-
[37]
Quantifying chemical short-range order in metallic alloys
Killian Sheriff, Yifan Cao, Tess Smidt, and Rodrigo Freitas. Quantifying chemical short-range order in metallic alloys. Proceedings of the National Academy of Sciences, 121: 0 e2322962121, 2024 b . doi:10.1073/pnas.2322962121
-
[38]
A. Tamm, A. Aabloo, M. Klintenberg, M. Stocks, and A. Caro. Atomic-scale properties of Ni-based FCC ternary, and quaternary alloys. Acta Materialia, 99: 0 307--312, 2015. doi:10.1016/j.actamat.2015.08.015
-
[39]
A. van de Walle, M. Asta, and G. Ceder. The alloy theoretic automated toolkit: A user guide. CALPHAD, 26 0 (4): 0 539--553, 2002. doi:10.1016/S0364-5916(02)80006-2
-
[40]
Flynn Walsh, Mark Asta, and Robert O. Ritchie. Magnetically driven short-range order can explain anomalous measurements in CrCoNi . Proceedings of the National Academy of Sciences, 118 0 (13): 0 e2020540118, 2021. doi:10.1073/pnas.2020540118
-
[41]
Flynn Walsh, Jun Zhang, Robert O. Ritchie, Andrew M. Minor, and Mark Asta. Ubiquitous short-range order in multi-principal element alloys. Nature Communications, 15: 0 6602, 2024. doi:10.1038/s41467-024-49606-1
-
[42]
B. E. Warren. X-ray diffraction study of the structure of alloys. Physical Review, 78: 0 821, 1950. doi:10.1103/PhysRev.78.821
-
[43]
Short-range ordering and its effects on mechanical properties of high-entropy alloys
Yuan Wu, Fei Zhang, Xiaoyuan Yuan, Hailong Huang, Xiaocan Wen, Yihan Wang, Mengyuan Zhang, Honghui Wu, Xiongjun Liu, Hui Wang, Suihe Jiang, and Zhaoping Lu. Short-range ordering and its effects on mechanical properties of high-entropy alloys. Journal of Materials Science & Technology, 62: 0 214--220, 2021 a . doi:10.1016/j.jmst.2020.06.018
-
[44]
Zhenggang Wu, Hongbin Bei, George M. Pharr, and Easo P. George. Recovery, recrystallization, grain growth and phase stability of a family of FCC -structured multi-component equiatomic solid solution alloys. Intermetallics, 46: 0 131--140, 2014. doi:10.1016/j.intermet.2013.10.024
-
[45]
Deformation mechanisms of high-entropy alloys revealed by in-situ studies
Zhengxiong Wu, Yongjie Zhao, Haibo Yang, Yu Song, Jinghua Qin, and En Ma. Deformation mechanisms of high-entropy alloys revealed by in-situ studies. Acta Materialia, 215: 0 117058, 2021 b . doi:10.1016/j.actamat.2021.117058
-
[46]
J.-W. Yeh, S.-K. Chen, S.-J. Lin, J.-Y. Gan, T.-S. Chin, T.-T. Shun, C.-H. Tsau, and S.-Y. Chang. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Advanced Engineering Materials, 6: 0 299--303, 2004. doi:10.1002/adem.200300567
-
[47]
Ruopeng Zhang, Shiteng Zhao, Jun Ding, Yan Chong, Tao Jia, Colin Ophus, Mark Asta, Robert O. Ritchie, and Andrew M. Minor. Short-range order and its impact on the CrCoNi medium-entropy alloy. Nature, 581: 0 283--287, 2020. doi:10.1038/s41586-020-2275-z
-
[48]
Atomic-scale evidence of chemical short-range order in CrCoNi medium-entropy alloy
Lingling Zhou, Qin Wang, Jian Wang, Xuefei Chen, Ping Jiang, Hao Zhou, Fuping Yuan, Xiaolei Wu, Zhiying Cheng, and En Ma. Atomic-scale evidence of chemical short-range order in CrCoNi medium-entropy alloy. Acta Materialia, 224: 0 117490, 2022. doi:10.1016/j.actamat.2021.117490
-
[49]
A. Zunger, S.-H. Wei, L. G. Ferreira, and J. E. Bernard. Special quasirandom structures. Physical Review Letters, 65: 0 353--356, 1990. doi:10.1103/PhysRevLett.65.353
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.