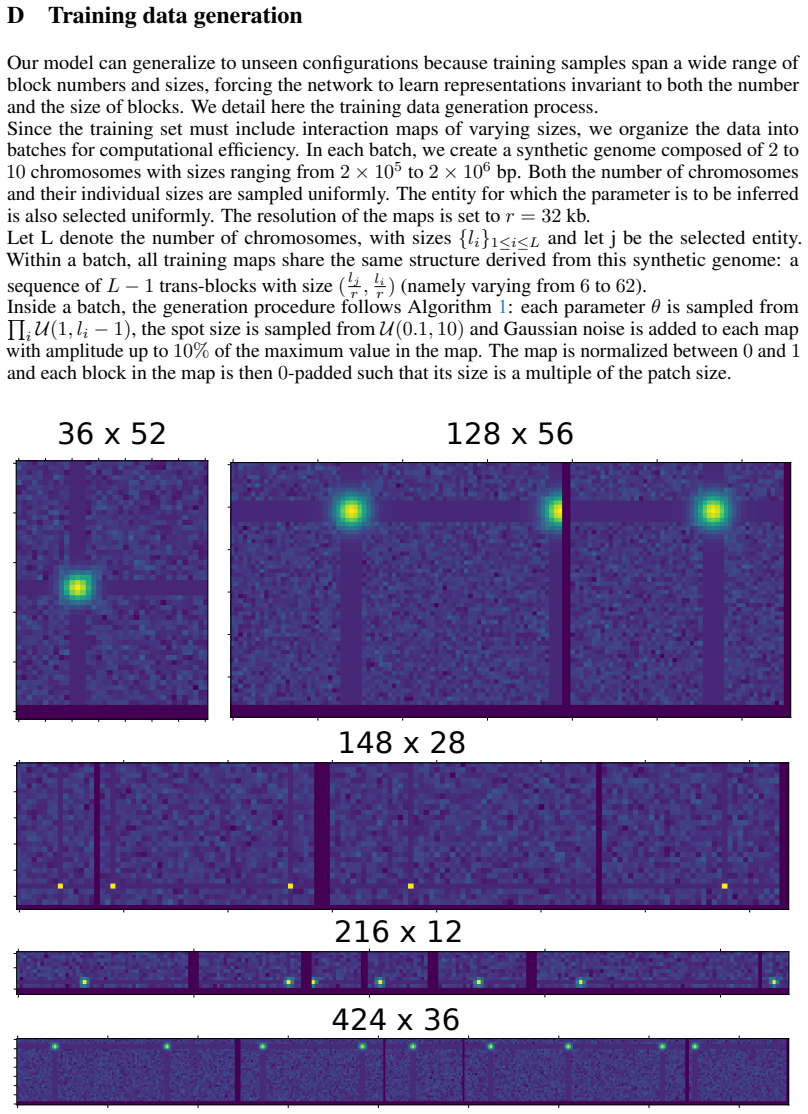
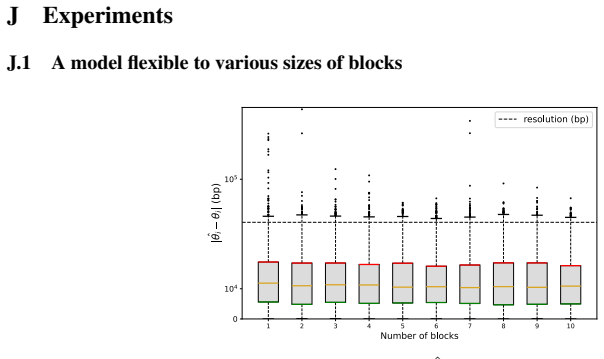
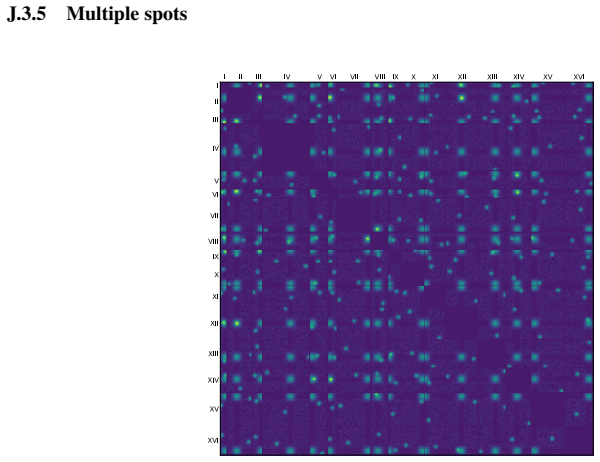
textit{BlockFormer} : Transformer-based inference from interaction maps
Pith reviewed 2026-05-22 09:11 UTC · model grok-4.3
The pith
A transformer trained on synthetic data accurately locates centromeres from Hi-C interaction maps across many species.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors claim that their BlockFormer transformer architecture, trained on abundant synthetic interaction maps, can accurately recover the genomic positions of centromeres from real Hi-C data across a wide range of species with varying genome sizes.
What carries the argument
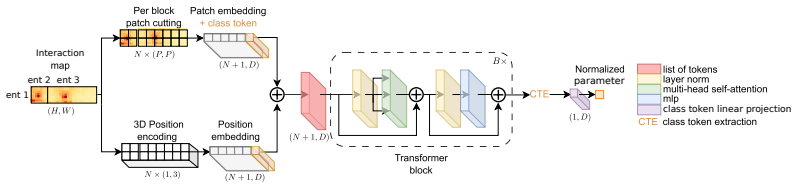
The BlockFormer transformer that processes interaction maps with variable entity numbers and sizes to infer localized parameters like centromere positions.
If this is right
- Accurate centromere position recovery in species with different genome sizes.
- Generalization to other inverse problems involving interaction maps.
- Reduced reliance on manual annotation or per-species models for genomic feature detection.
- Scalable analysis using computationally cheap synthetic training data.
Where Pith is reading between the lines
- Similar methods could apply to other pairwise interaction data in biology or physics.
- Training on synthetic data might reduce the need for large real datasets in genomic inference tasks.
- Extensions could handle even more variable or noisy interaction maps from different experimental techniques.
Load-bearing premise
Interaction maps from different species share enough common structure, such as aligned localized patterns, for a single transformer model to handle the variability in block numbers and sizes.
What would settle it
The model producing inaccurate centromere positions when tested on Hi-C maps from a species whose interaction patterns deviate substantially from those in the synthetic training data.
Figures





















read the original abstract
Inference from interaction maps, such as centromere identification from genome-wide chromosome conformation capture techniques -- notably Hi-C -- can be formulated as a generic inverse problem: infer a set of parameters given a map summarizing pairwise interactions between entities through blocks of variable numbers and sizes. In this work, we introduce a data-driven approach that leverages shared structure between these maps, such as global alignment between localized patterns, while handling the variability in number and size of entities arising in real-world data. Our approach relies on a transformer architecture capable of handling such variability and a custom simulator to generate abundant, yet computationally cheap synthetic data for training. Applied to the problem of centromere localization, the method accurately recovers their genomic positions across a wide range of species of various genome sizes.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces BlockFormer, a transformer architecture for inferring a set of parameters from interaction maps that share global alignment of localized patterns but exhibit variability in the number and size of entities. Training relies on synthetic data from a custom simulator; the method is applied to centromere localization in Hi-C maps and claims accurate recovery of genomic positions across species with varying genome sizes.
Significance. If the simulator-to-real generalization is substantiated, the work provides a scalable, data-driven alternative to traditional inverse-problem methods for interaction maps, leveraging transformer flexibility for variable-length inputs without fixed entity assumptions. This could benefit automated analysis in genomics and related domains where interaction matrices arise.
major comments (2)
- Abstract: the claim that the method 'accurately recovers' centromere genomic positions across species supplies no quantitative metrics, baseline comparisons, error bars, or validation details, leaving the central empirical claim unsupported on the information given.
- Simulator and training description (Methods section): the custom simulator used to generate training interaction maps is not shown to reproduce key statistical features of real Hi-C data (contact decay, compartment biases, noise structure, variable block sizes); without such fidelity checks the simulator-to-real transfer that underpins the generalization claim remains unvalidated.
minor comments (2)
- Clarify the precise tokenization and positional encoding scheme used to accommodate variable numbers of blocks within the transformer input.
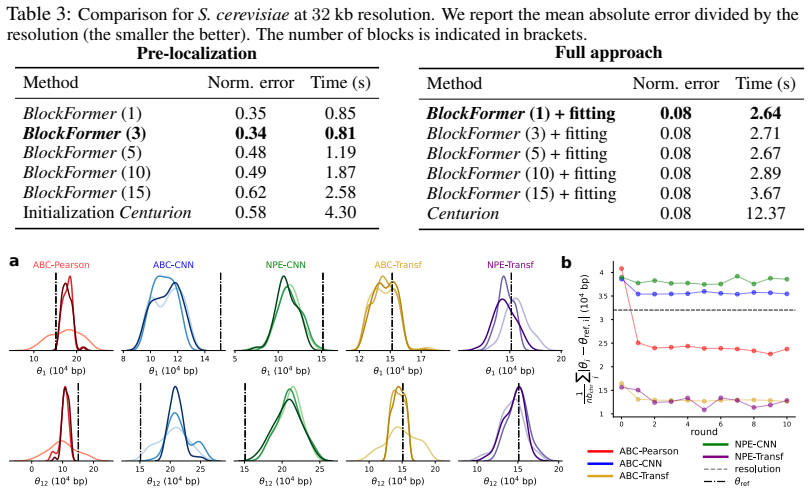
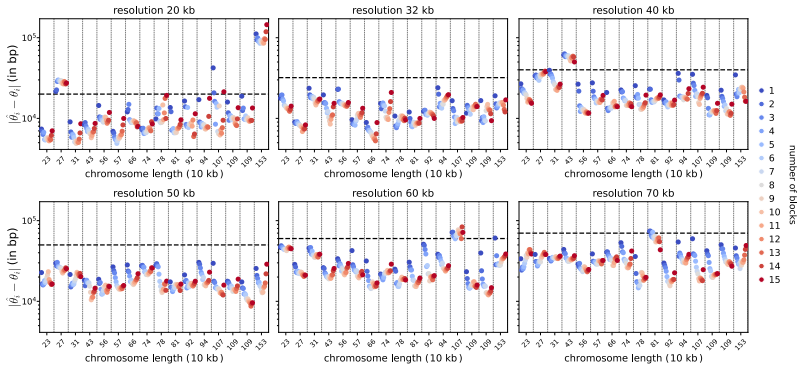
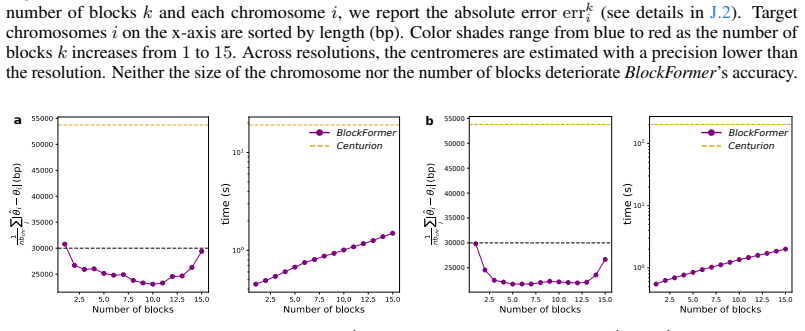
- Figure captions and axis labels should explicitly state the species, genome sizes, and quantitative recovery metrics shown.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback, which has helped us identify areas where the manuscript can be strengthened. We address each major comment below and outline the revisions we will make.
read point-by-point responses
-
Referee: Abstract: the claim that the method 'accurately recovers' centromere genomic positions across species supplies no quantitative metrics, baseline comparisons, error bars, or validation details, leaving the central empirical claim unsupported on the information given.
Authors: We agree that the abstract's claim would be better supported by quantitative evidence. The original abstract was kept concise to highlight the overall contribution, but this omitted key details from our experiments. In the revised manuscript we will expand the abstract to include specific performance metrics (e.g., mean absolute error in base-pair localization with standard deviation across species) and a brief reference to baseline comparisons performed in the results section. revision: yes
-
Referee: Simulator and training description (Methods section): the custom simulator used to generate training interaction maps is not shown to reproduce key statistical features of real Hi-C data (contact decay, compartment biases, noise structure, variable block sizes); without such fidelity checks the simulator-to-real transfer that underpins the generalization claim remains unvalidated.
Authors: The referee is correct that explicit fidelity validation was not included in the submitted version. While the simulator was designed to capture the block-like interaction patterns and length variability central to centromere localization, we did not present direct statistical comparisons (such as contact decay curves or block-size histograms) against real Hi-C data. We will add a dedicated subsection to the Methods (or supplementary material) that quantifies these matches, including plots and metrics for contact probability decay, noise levels, and block-size distributions between simulated and real maps from the evaluated species. revision: yes
Circularity Check
No circularity: training on independent custom simulator with real-data evaluation
full rationale
The paper formulates centromere localization as an inverse problem and solves it via a transformer trained exclusively on synthetic interaction maps produced by a custom simulator, then evaluated on real Hi-C data from multiple species. No equations or claims reduce a prediction to a fitted parameter on the same data, no self-definitional loops appear, and no load-bearing self-citations or uniqueness theorems are invoked to force the architecture or results. The simulator-to-real transfer is an external generalization step rather than a tautological renaming or fit; the derivation chain therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Our approach relies on a transformer architecture capable of handling such variability and a custom simulator to generate abundant, yet computationally cheap synthetic data for training.
-
IndisputableMonolith/Foundation/AlexanderDuality.leanalexander_duality_circle_linking unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
per-block 3D positional encoding … (i, j, k) where i is the block index
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.