When Does Structure Help? The Information Bonus of AlphaFold2 Representations over Protein Language Models
Pith reviewed 2026-06-28 07:39 UTC · model grok-4.3
The pith
AlphaFold2 representations outperform sequence models only on allostery by capturing long-range geometry.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
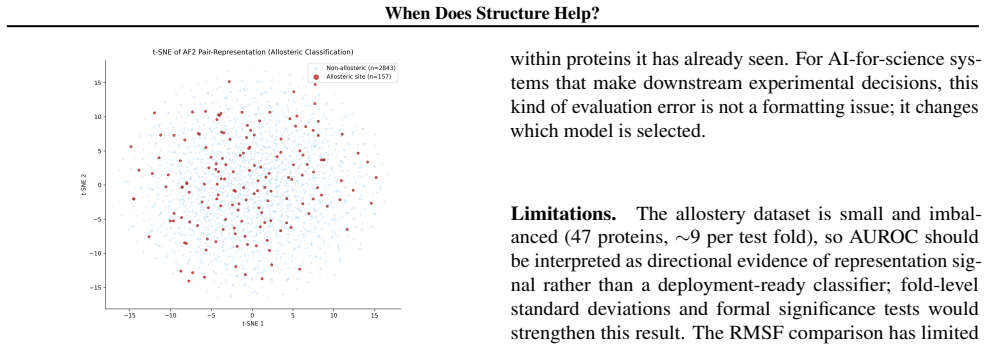
The central claim is that the information bonus of AlphaFold2 Evoformer representations over ESM-2 embeddings is sharply mechanism-dependent: it is negative for binding affinity (IB = -0.141) and binary flexibility (IB = -0.060), but positive for allosteric-site classification (IB = +0.064), where only the AF2 representations produce above-chance predictions because they alone encode long-range geometric signal absent from sequence alone.
What carries the argument
The information bonus (IB), a task-level metric that measures the linearly accessible advantage of frozen single-sequence AlphaFold2 Evoformer representations over frozen ESM-2 embeddings under protein-level cross-validation.
If this is right
- For allosteric-site tasks, pipelines should default to AF2-derived representations rather than sequence-only models.
- For binding-affinity and flexibility tasks, sequence-only models can be used without loss of linear performance and at lower cost.
- Any evaluation that splits at the residue level must be replaced by protein-level splits to avoid leakage that can reverse representation rankings.
- Representation selection in AI-for-science systems becomes a measurable, task-specific decision rather than a default choice.
Where Pith is reading between the lines
- Allostery appears to depend on global 3-D geometry that local sequence statistics cannot recover.
- The same information-bonus test could be applied to other long-range tasks such as protein-protein interface prediction or conformational change classification.
- If the leakage artifact is general, many published residue-level benchmarks for protein representations may need re-evaluation.
Load-bearing premise
That linear probes together with protein-level cross-validation fully isolate the contribution of each representation without residual task-specific biases or unaccounted data leakage.
What would settle it
A re-run of the allostery task that uses a different probe architecture or stricter protein-level splits and finds that the positive information bonus disappears or reverses sign.
Figures



read the original abstract
AI scientist systems increasingly choose biological foundation models before they choose experiments. In protein pipelines, this creates a concrete engineering and scientific question: when is the cost of structural inference worth paying over a cheaper sequence-only model? We introduce the information bonus (IB), a task-level metric that measures the linearly accessible advantage of frozen single-sequence AlphaFold2 Evoformer representations over frozen ESM-2 embeddings under protein-level cross-validation. Across binding affinity regression (PDBbind, n=5,680), conformational flexibility (ATLAS molecular dynamics, 268 proteins), and allosteric-site classification (AlloSigDB, n=9,925 residues), IB is sharply mechanism-dependent. ESM-2 dominates binding affinity (IB=-0.141; Pearson r=0.449 vs. 0.307) and binary flexibility (IB=-0.060; AUROC 0.824 vs. 0.764; p=0.0017). AF2 single representations give the only above-chance allostery predictions (IB=+0.064; AUROC 0.548 vs. 0.485), revealing long-range geometric signal not recovered from sequence alone. We also identify a residue-level leakage artifact: naive residue splits inflate RMSF performance by 27-39% depending on the representation, enough to reverse representation rankings. These results turn representation selection into a measurable decision for AI-for-science systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces the 'information bonus' (IB) metric to quantify the linearly accessible advantage of frozen AlphaFold2 Evoformer single representations over ESM-2 embeddings under protein-level cross-validation. It evaluates this on binding affinity regression (PDBbind), conformational flexibility (ATLAS), and allosteric site classification (AlloSigDB), finding ESM-2 superior for the first two tasks (IB negative) and AF2 superior for allostery (IB = +0.064, only above-chance AUROC), while also documenting a residue-level data leakage artifact in cross-validation splits.
Significance. If the central claims hold, the work offers a concrete, task-dependent decision criterion for choosing between sequence and structure foundation models in protein pipelines, with the allostery result suggesting unique long-range geometric information in AF2 representations. The identification of the residue-split leakage artifact (inflating RMSF performance by 27-39%) is a broadly applicable caution that strengthens the paper's utility for the field.
major comments (2)
- [Abstract (allostery results)] The claim that AF2 representations provide the only above-chance allostery predictions (IB=+0.064; AUROC 0.548 vs. 0.485) depends on the protein-level cross-validation (9,925 residues) fully isolating representation contributions without residual leakage from homologous proteins, shared MSAs, or overlap with AF2 training structures. Although the paper correctly identifies analogous leakage for residue splits on RMSF, equivalent validation is not shown for the allostery split, which is load-bearing for attributing the positive IB to geometric signal absent from sequence embeddings.
- [Abstract / Methods description] The abstract reports concrete performance numbers and a p-value (0.0017) but provides no details on representation extraction from the AF2 Evoformer, linear probe training, or the precise construction of the protein-level cross-validation splits. This lack of methodological transparency is load-bearing for reproducing and assessing the soundness of the IB values across tasks.
minor comments (1)
- The definition and computation of the Information Bonus (IB) could be clarified with an explicit formula or pseudocode to aid interpretation of the signed values.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback emphasizing methodological rigor and validation of cross-validation splits. We address each major comment below and will revise the manuscript to improve transparency and provide the requested checks.
read point-by-point responses
-
Referee: [Abstract (allostery results)] The claim that AF2 representations provide the only above-chance allostery predictions (IB=+0.064; AUROC 0.548 vs. 0.485) depends on the protein-level cross-validation (9,925 residues) fully isolating representation contributions without residual leakage from homologous proteins, shared MSAs, or overlap with AF2 training structures. Although the paper correctly identifies analogous leakage for residue splits on RMSF, equivalent validation is not shown for the allostery split, which is load-bearing for attributing the positive IB to geometric signal absent from sequence embeddings.
Authors: We agree that explicit validation of the allostery protein-level splits is necessary to support the attribution of the positive IB. The manuscript already shows the residue-split leakage effect for RMSF but did not include equivalent checks for AlloSigDB. In revision we will add analyses confirming no sequence homology (e.g., via BLAST or MMseqs2) and no shared MSAs between train and test proteins in the allostery splits. For potential overlap with AF2 training structures we will add a discussion of the implications and, where feasible, report the fraction of AlloSigDB proteins that appear in the public AF2 training set description; we note that any such overlap would be task-specific and does not affect the comparative IB metric between the two frozen models. revision: yes
-
Referee: [Abstract / Methods description] The abstract reports concrete performance numbers and a p-value (0.0017) but provides no details on representation extraction from the AF2 Evoformer, linear probe training, or the precise construction of the protein-level cross-validation splits. This lack of methodological transparency is load-bearing for reproducing and assessing the soundness of the IB values across tasks.
Authors: We acknowledge that the abstract is concise and that the current Methods section could be expanded for full reproducibility. In the revised manuscript we will (1) add a brief clause in the abstract summarizing the representation source (AF2 Evoformer single representation), probe type (linear model with L2 regularization), and split protocol (protein-level random split with no intra-protein residue leakage), and (2) expand the Methods section with explicit details on extraction (which layer and which tensor), hyperparameter selection for the linear probes, exact criteria used to construct the protein-level CV folds, and code repository link. These changes will make the reported IB values and p-value fully reproducible without altering any numerical results. revision: yes
Circularity Check
No circularity: empirical head-to-head metrics on held-out proteins
full rationale
The paper defines the information bonus (IB) as the measured performance difference between linear probes on frozen AF2 vs. ESM-2 representations under protein-level cross-validation. All reported values (e.g., IB=+0.064 for allostery AUROC) are computed directly from independent held-out evaluations on PDBbind, ATLAS, and AlloSigDB splits. No equations, self-citations, or ansatzes reduce these differences to fitted parameters or prior results by construction. The paper explicitly flags and quantifies the residue-split leakage artifact rather than concealing it. The derivation chain is therefore self-contained empirical measurement.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Linear probes extract the relevant information from the frozen embeddings
invented entities (1)
-
Information Bonus (IB)
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Highly accurate protein structure prediction with
Jumper, John and Evans, Richard and Pritzel, Alexander and Green, Tim and Figurnov, Michael and Ronneberger, Olaf and Tunyasuvunakool, Kathryn and Bates, Russ and Zidek, Augustin and Potapenko, Anna and others , journal=. Highly accurate protein structure prediction with
-
[2]
Science , volume=
Evolutionary-scale prediction of atomic-level protein structure with a language model , author=. Science , volume=
-
[3]
Proceedings of the National Academy of Sciences , volume=
Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences , author=. Proceedings of the National Academy of Sciences , volume=
-
[4]
Elnaggar, Ahmed and Heinzinger, Michael and Dallago, Christian and Rehawi, Ghalia and Wang, Yu and Jones, Llion and Gibbs, Tom and Feher, Tamas and Angerer, Christoph and Steinegger, Martin and others , journal=
-
[5]
Su, Jin and Han, Chenchen and Zhou, Yuyang and Shan, Junjie and Zhou, Xibin and Yuan, Fajie , booktitle=
-
[6]
Nature Methods , volume=
Mirdita, Milot and Sch. Nature Methods , volume=
-
[7]
, journal=
Rao, Roshan and Bhattacharya, Nicholas and Thomas, Neil and Duan, Yan and Chen, Peter and Canny, John and Abbeel, Pieter and Song, Yun S. , journal=. Evaluating protein transfer learning with
-
[8]
arXiv preprint arXiv:1610.01644 , year=
Understanding intermediate layers using linear classifier probes , author=. arXiv preprint arXiv:1610.01644 , year=
-
[9]
and Xiong, Caiming and Socher, Richard and Rajani, Nazneen Fatema , booktitle=
Vig, Jesse and Madani, Ali and Varshney, Lav R. and Xiong, Caiming and Socher, Richard and Rajani, Nazneen Fatema , booktitle=
-
[10]
Advances in Neural Information Processing Systems , volume=
Language models enable zero-shot prediction of the effects of mutations on protein function , author=. Advances in Neural Information Processing Systems , volume=
-
[11]
Cell Systems , volume=
Evolutionary velocity with protein language models predicts evolutionary dynamics of diverse proteins , author=. Cell Systems , volume=
-
[12]
Proceedings of the National Academy of Sciences , volume=
Sparse autoencoders uncover biologically interpretable features in protein language model representations , author=. Proceedings of the National Academy of Sciences , volume=. 2025 , doi=
2025
-
[13]
2025 , doi=
Simon, Elana and Zou, James , journal=. 2025 , doi=
2025
-
[14]
Vander Meersche, Yann and Cretin, Guillaume and Goncearenco, Alexander and Sterckx, Yann G. J. and Gelly, Jean-Christophe and de Brevern, Alexandre G. , journal=
-
[15]
Liu, Zhihai and Li, Yan and Han, Lixia and Li, Jie and Liu, Jian and Zhao, Zhong and Nie, Wenkai and Liu, Yuyang and Wang, Renxiao , journal=
-
[16]
and others , journal=
Li, Lijun and Chang, Shirley and Bhaskara, Ramachandra M. and others , journal=
-
[17]
and Bichmann, Lorenz and Keedy, Daniel A
Cimermancic, Peter and Weinkam, Patrick and Rettenmaier, Thomas J. and Bichmann, Lorenz and Keedy, Daniel A. and Woldeyes, Rahel A. and Schneidman-Duhovny, Dina and Demerdash, Omar N. and Mitchell, Julie C. and Wells, James A. and others , journal=
-
[18]
Journal of Chemical Information and Modeling , volume=
Analyzing learned molecular representations for property prediction , author=. Journal of Chemical Information and Modeling , volume=
-
[19]
Varadi, Mihaly and Anyango, Stephen and Deshpande, Mandar and Nair, Sreenath and Natassia, Christine and Yordanova, Galabina and Yuan, David and Stroe, Oana and Wood, Gemma and Laydon, Agata and others , journal=
-
[20]
Lu, Chris and Lu, Cong and Lange, Robert Tjarko and Foerster, Jakob and Clune, Jeff and Ha, David , journal=. The
-
[21]
Briefings in Bioinformatics , volume=
Protein language models are performant in structure-free virtual screening , author=. Briefings in Bioinformatics , volume=. 2024 , doi=
2024
-
[22]
Piao, Haixing and others , journal=
-
[23]
PLOS ONE , volume=
The role of data imbalance bias in the prediction of protein stability change upon mutation , author=. PLOS ONE , volume=. 2023 , doi=
2023
-
[24]
ICLR 2024 Workshop on Generative and Experimental Perspectives for Biomolecular Design , year=
Revealing data leakage in protein interaction benchmarks , author=. ICLR 2024 Workshop on Generative and Experimental Perspectives for Biomolecular Design , year=
2024
-
[25]
Briefings in Bioinformatics , volume=
Cracking the black box of deep sequence-based protein-protein interaction prediction , author=. Briefings in Bioinformatics , volume=. 2024 , doi=
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.