Balancing label resolution and computational cost in dynamical models of lipid metabolism
Pith reviewed 2026-06-27 04:40 UTC · model grok-4.3
The pith
Modelling three of five labels balances inferential power and computational cost in lipid metabolism models.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
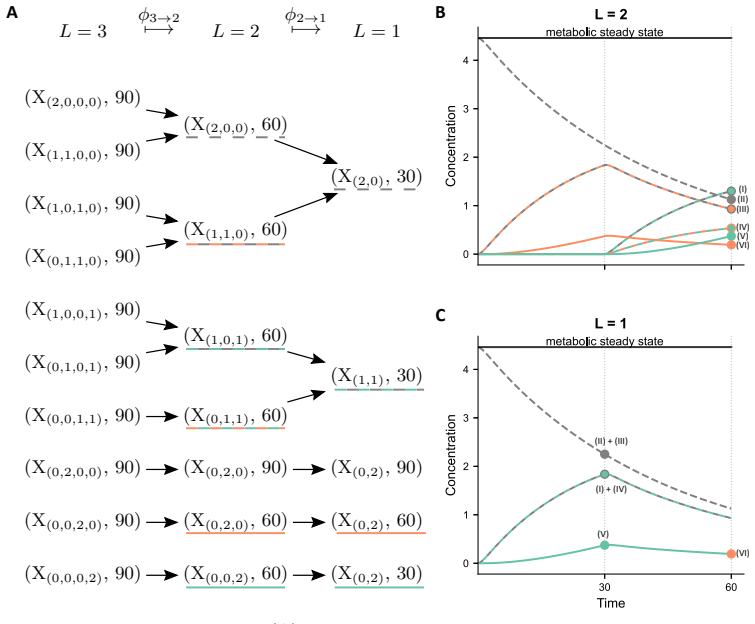
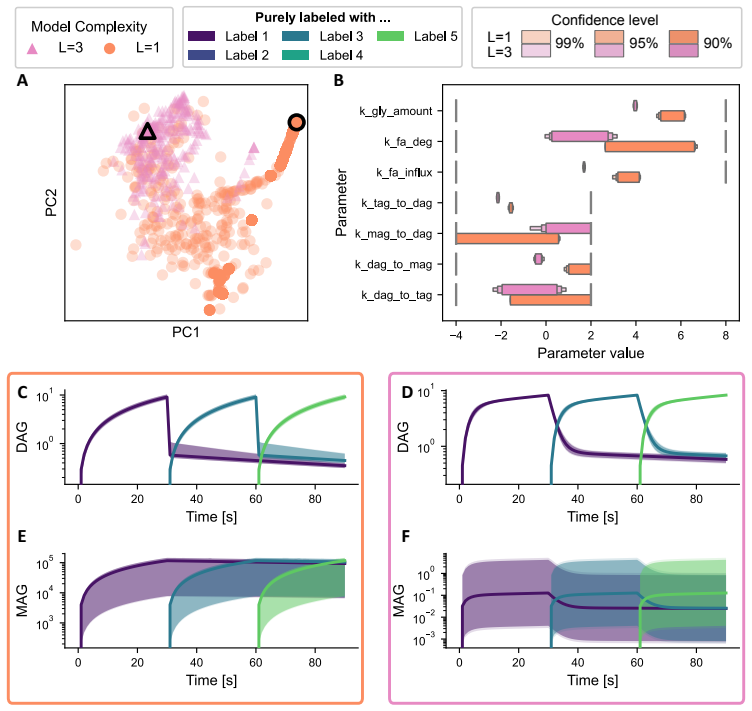
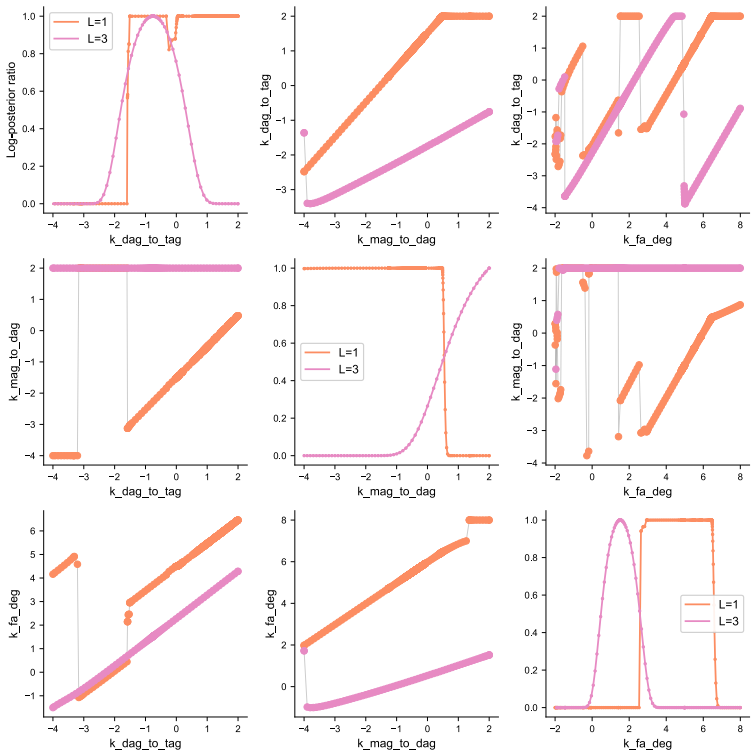
The authors establish that in multi-label experiments for lipid metabolism, models resolving three out of five labels achieve a practical balance between experimental feasibility, inferential power, and computational tractability. Single-label models can produce biologically implausible predictions for latent variables, while higher resolution models better constrain these dynamics.
What carries the argument
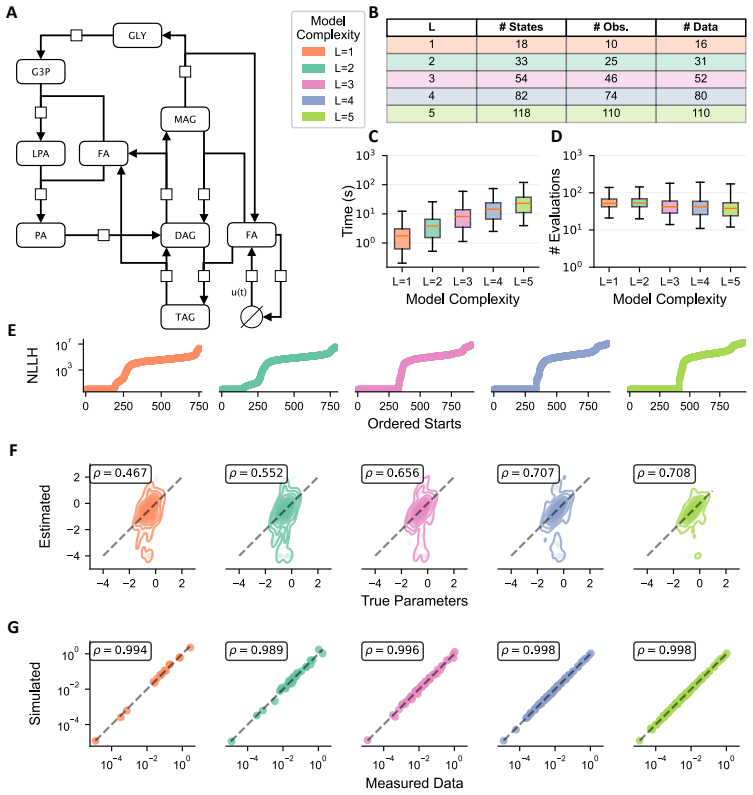
The dynamical model of lipid metabolism parameterized by the number of modelled labels, where increasing labels expands the state space and thus the computational burden of parameter estimation.
If this is right
- Models using three labels maintain high accuracy in estimating metabolic parameters.
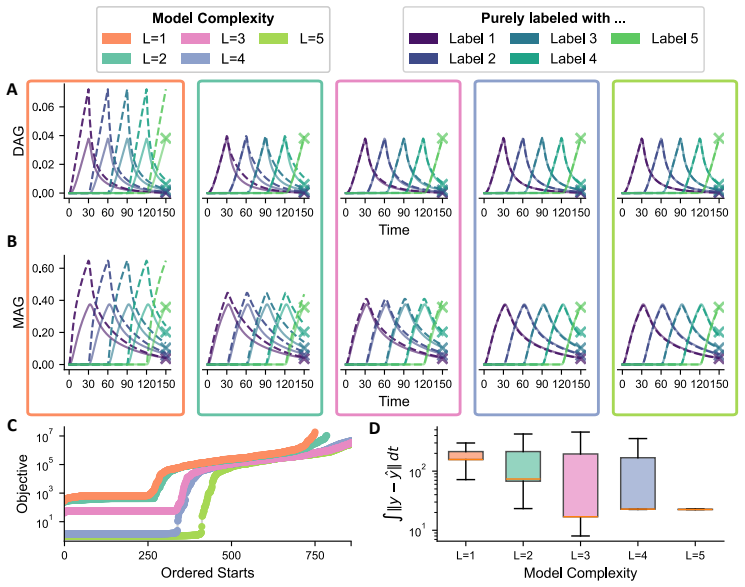
- Recovery of temporal trajectories improves with additional labels but with diminishing returns.
- Computational cost rises rapidly as more labels are included in the model.
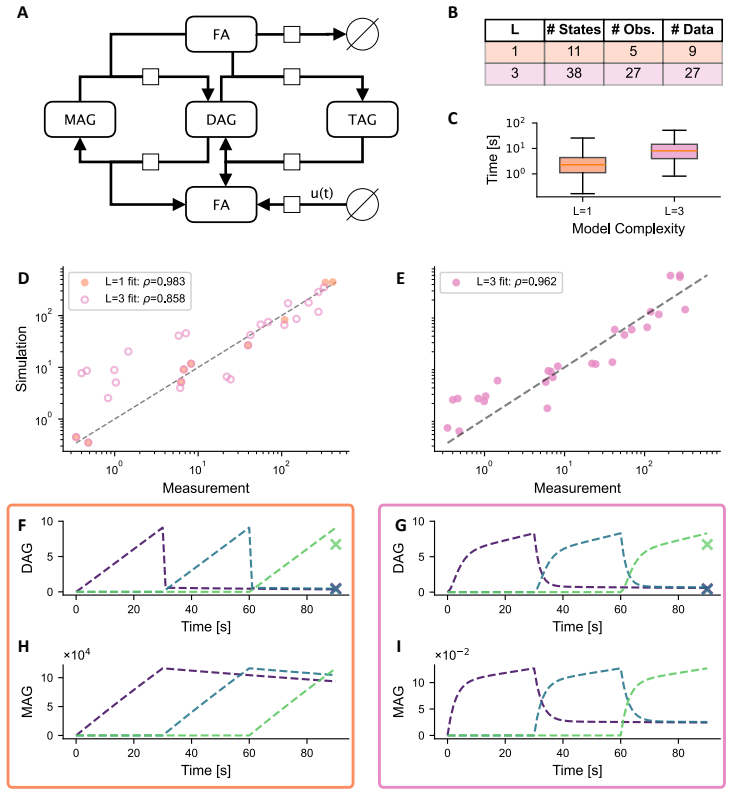
- Application to triglyceride cycling demonstrates that resolving more labels prevents implausible predictions for unobserved lipid species.
Where Pith is reading between the lines
- Similar label-resolution trade-offs may apply to other multi-isotope experiments in systems biology.
- Validation on real experimental data would confirm if synthetic performance rankings hold.
- The approach could guide label selection in related metabolic modelling studies.
Load-bearing premise
The synthetic data generated from the five-label experiment accurately captures the statistical properties and identifiability structure of real experimental measurements.
What would settle it
An experiment where real five-label mass spectrometry data is collected and used to compare the predictive performance of three-label versus five-label models on held-out measurements.
Figures
read the original abstract
Lipid metabolism is a central biological process that is commonly studied using destructive mass-spectrometry experiments. A recently proposed strategy, uses multiple labels to extract temporal information about lipid metabolism from a single destructive measurement. However, the computational complexity of the model-based data analysis increases rapidly with the number of labels, creating a fundamental trade-off between the information content of the measurements and the cost of analysis. Here, we examine how the number of modelled labels affects parameter estimation accuracy, trajectory recovery, and computational cost, and whether modelling fewer labels than are experimentally available can mitigate this trade-off. Using synthetic data from a five-label experiment, we find that modelling three of the five labels provides a practical balance between experimental feasibility, inferential power, and computational tractability. In an application to hepatocyte triglyceride cycling, we further show that the most cost-efficient, single-label model can yield biologically implausible predictions for unobserved species, whereas models that resolve more labels better constrain these latent dynamics. These results provide practical guidance for selecting model resolution in multi-label experiments and establish a quantitative basis for balancing inferential power against computational cost.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript examines the trade-off between label resolution and computational cost in dynamical models of lipid metabolism inferred from destructive mass-spectrometry data. It uses synthetic data generated from a five-label model to compare parameter estimation accuracy, trajectory recovery, and runtime across different numbers of modeled labels, concluding that three labels provide a practical balance of experimental feasibility, inferential power, and tractability. A hepatocyte triglyceride-cycling application is presented to show that single-label models can produce biologically implausible latent trajectories while higher-resolution models better constrain them.
Significance. If the reported ranking of label counts is robust, the work supplies concrete, quantitative guidance for experimental design in multi-label lipid studies, a domain where destructive sampling makes temporal resolution expensive. The combination of controlled synthetic benchmarks with a real biological case is a positive feature; however, the strength depends on whether the synthetic-data ranking generalizes.
major comments (2)
- [Synthetic-data experiments and hepatocyte application] The central claim that three labels strike the optimal balance rests entirely on performance rankings obtained from synthetic data generated under the exact five-label dynamical model (with controlled noise). This perfect model match leaves open whether the ranking would change under realistic misspecification such as unmodeled reactions, correlated measurement errors, or non-Gaussian noise; the hepatocyte case only illustrates qualitative differences and does not quantitatively re-rank resolutions on actual observations.
- [Results on synthetic data] No uncertainty quantification (e.g., posterior widths or bootstrap intervals) is reported for the trajectory-recovery or parameter-estimation metrics that underpin the three-label recommendation; without these, it is difficult to judge whether the reported advantage of three versus two or four labels is statistically distinguishable from sampling variability.
minor comments (1)
- [Abstract] The abstract states that the synthetic data come from a 'five-label experiment' but does not specify whether the generative model includes all reactions or only a subset; clarifying this would help readers assess the scope of the identifiability claims.
Simulated Author's Rebuttal
Thank you for the detailed review. We appreciate the points raised about the synthetic data setup and the lack of uncertainty measures. Below we respond point by point to the major comments.
read point-by-point responses
-
Referee: [Synthetic-data experiments and hepatocyte application] The central claim that three labels strike the optimal balance rests entirely on performance rankings obtained from synthetic data generated under the exact five-label dynamical model (with controlled noise). This perfect model match leaves open whether the ranking would change under realistic misspecification such as unmodeled reactions, correlated measurement errors, or non-Gaussian noise; the hepatocyte case only illustrates qualitative differences and does not quantitatively re-rank resolutions on actual observations.
Authors: We designed the synthetic experiments to provide a controlled benchmark where the data-generating process is known exactly, allowing us to isolate the effects of label resolution on estimation accuracy and trajectory recovery without confounding factors from model mismatch. This is a standard approach in methodological studies to establish baseline performance. We acknowledge that real-world data may involve misspecification, and the hepatocyte application serves to illustrate that even on experimental data, single-label models can produce implausible latent trajectories while higher-resolution models constrain them better. However, without ground-truth trajectories in the real data, a quantitative re-ranking is not feasible. We will add a discussion of these limitations and potential impacts of misspecification in the revised manuscript. revision: partial
-
Referee: [Results on synthetic data] No uncertainty quantification (e.g., posterior widths or bootstrap intervals) is reported for the trajectory-recovery or parameter-estimation metrics that underpin the three-label recommendation; without these, it is difficult to judge whether the reported advantage of three versus two or four labels is statistically distinguishable from sampling variability.
Authors: The reported metrics are computed as averages over multiple independent synthetic datasets for each label resolution, providing some indication of robustness. Nevertheless, we agree that explicit uncertainty quantification, such as standard errors or bootstrap intervals on the performance metrics, would strengthen the comparison. We will revise the results section to include measures of variability across the simulation replicates to allow assessment of statistical distinguishability. revision: yes
Circularity Check
No significant circularity; evaluation uses independent synthetic benchmarks
full rationale
The paper generates synthetic data once from the five-label dynamical model and then computes identifiability, trajectory recovery, and cost metrics separately for each reduced label count (1-5). These metrics are not fitted parameters renamed as predictions, nor are any load-bearing claims justified solely by self-citation. The central recommendation for three labels follows directly from the tabulated simulation outcomes rather than from any definitional equivalence or ansatz smuggled via prior work. The derivation chain therefore remains self-contained against the external synthetic benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Systems Biology: A Brief Overview
Kitano H. Systems Biology: A Brief Overview. Science. 2002 03;295(5560):1662-4. doi:10.1126/science.1069492
-
[2]
de Jong H. Modeling and Simulation of Genetic Regulatory Systems: A Literature Review. Journal of Computational Biology. 2002 2026/04/09;9(1):67-103. doi:10.1089/10665270252833208
-
[3]
Physicochemical modelling of cell signalling pathways
Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK. Physicochemical modelling of cell signalling pathways. Nat Cell Biol. 2006 11;8(11):1195-203. doi:10.1038/ncb1497
-
[4]
Dynamic models for metabolomics data integration
Lakrisenko P, Weindl D. Dynamic models for metabolomics data integration. Current Opinion in Systems Biology. 2021:100358. doi:https://doi.org/10.1016/j.coisb.2021.100358
-
[5]
Identifiability and observability analysis for experimental design in nonlinear dynamical models
Raue A, Becker V, Klingm¨ uller U, Timmer J. Identifiability and observability analysis for experimental design in nonlinear dynamical models. Chaos. 2010 Dec;20(045105). doi:10.1063/1.3528102
-
[6]
On structural and practical identifiability
Wieland FG, Hauber AL, Rosenblatt M, T¨ onsing C, Timmer J. On structural and practical identifiability. Current Opinion in Systems Biology. 2021;25:60-9. doi:10.1016/j.coisb.2021.03.005
-
[7]
RNA-Seq methods for transcriptome analysis
Hrdlickova R, Toloue M, Tian B. RNA-Seq methods for transcriptome analysis. WIREs RNA. 2017 2026/02/26;8(1):e1364. doi:https://doi.org/10.1002/wrna.1364
-
[8]
A Review on Macroscale and Microscale Cell Lysis Methods
Shehadul Islam M, Aryasomayajula A, Selvaganapathy PR. A Review on Macroscale and Microscale Cell Lysis Methods. Micromachines. 2017;8(3):83. doi:10.3390/mi8030083. June 12, 2026 19/21
-
[9]
Liquid chromatography–tandem mass spectrometry for clinical diagnostics
Thomas SN, French D, Jannetto PJ, Rappold BA, Clarke WA. Liquid chromatography–tandem mass spectrometry for clinical diagnostics. Nature Reviews Methods Primers. 2022;2(1):96. doi:10.1038/s43586-022-00175-x
-
[10]
Advances and Trends in Omics Technology Development
Dai X, Shen L. Advances and Trends in Omics Technology Development. Frontiers in Medicine. 2022;Volume 9 - 2022. doi:10.3389/fmed.2022.911861
-
[11]
An Introduction to Mass Spectrometry-Based Proteomics
Shuken SR. An Introduction to Mass Spectrometry-Based Proteomics. Journal of Proteome Research. 2023 07;22(7):2151-71. doi:10.1021/acs.jproteome.2c00838
-
[12]
Are batch effects still relevant in the age of big data? Trends in Biotechnology
Goh WWB, Yong CH, Wong L. Are batch effects still relevant in the age of big data? Trends in Biotechnology. 2022 09;40(9):1029-40. doi:10.1016/j.tibtech.2022.02.005
-
[13]
Tracing metabolic flux in vivo: basic model structures of tracer methodology
Kim IY, Park S, Kim Y, Kim HJ, Wolfe RR. Tracing metabolic flux in vivo: basic model structures of tracer methodology. Experimental & Molecular Medicine. 2022;54(9):1311-22. doi:10.1038/s12276-022-00814-z
-
[14]
Live-cell imaging powered by computation
Shroff H, Testa I, Jug F, Manley S. Live-cell imaging powered by computation. Nature Reviews Molecular Cell Biology. 2024;25(6):443-63. doi:10.1038/s41580-024-00702-6
-
[15]
Recent Advancement in NMR Based Plant Metabolomics: Techniques, Tools, and Analytical Approaches
Kumar N, Jaitak V. Recent Advancement in NMR Based Plant Metabolomics: Techniques, Tools, and Analytical Approaches. Critical Reviews in Analytical Chemistry. 2026 01;56(1):1-25. doi:10.1080/10408347.2024.2375314
-
[16]
Non-invasive Raman spectroscopy for time-resolved in-line lipidomics
Wieland K, Masri M, von Poschinger J, Br¨ uck T, Haisch C. Non-invasive Raman spectroscopy for time-resolved in-line lipidomics. RSC Advances. 2021;11(46):28565-72. doi:10.1039/D1RA04254H
-
[17]
Advantages and Pitfalls of Mass Spectrometry Based Metabolome Profiling in Systems Biology
Aretz I, Meierhofer D. Advantages and Pitfalls of Mass Spectrometry Based Metabolome Profiling in Systems Biology. International Journal of Molecular Sciences. 2016;17(5):632. doi:10.3390/ijms17050632
-
[18]
Analytical Considerations of Stable Isotope Labelling in Lipidomics
Triebl A, Wenk MR. Analytical Considerations of Stable Isotope Labelling in Lipidomics. Biomolecules. 2018;8(4):151. doi:10.3390/biom8040151
-
[19]
Jost PJ, Weindl D, Wunderling K, Thiele C, Hasenauer J. Pseudo-time trajectory of single-cell lipidomics: Suggestion for experimental setup and computational analysis. bioRxiv. 2025 01:2025.04.11.648323. doi:10.1101/2025.04.11.648323
-
[20]
BioNetGen 2.2: advances in rule-based modeling
Harris LA, Hogg JS, Tapia JJ, Sekar JAP, Gupta S, Korsunsky I, et al. BioNetGen 2.2: advances in rule-based modeling. Bioinformatics. 2016 07;32(21):3366-8. doi:10.1093/bioinformatics/btw469
-
[21]
The Kappa platform for rule-based modeling
Boutillier P, Maasha M, Li X, Medina-Abarca HF, Krivine J, Feret J, et al. The Kappa platform for rule-based modeling. Bioinformatics. 2018 06;34(13):i583-92. doi:10.1093/bioinformatics/bty272
-
[22]
Profile likelihood in systems biology
Kreutz C, Raue A, Kaschek D, Timmer J. Profile likelihood in systems biology. FEBS J. 2013;280(11):2564-71. doi:10.1111/febs.12276
-
[23]
PEtab—Interoperable specification of parameter estimation problems in systems biology
Schmiester L, Sch¨ alte Y, Bergmann FT, Camba T, Dudkin E, Egert J, et al. PEtab—Interoperable specification of parameter estimation problems in systems biology. PLoS Computational Biology. 2021 January;17(1):1-10. doi:10.1371/journal.pcbi.1008646. June 12, 2026 20/21
-
[24]
AMICI: high-performance sensitivity analysis for large ordinary differential equation models
Fr¨ ohlich F, Weindl D, Sch¨ alte Y, Pathirana D, Paszkowski L, Lines GT, et al. AMICI: high-performance sensitivity analysis for large ordinary differential equation models. Bioinformatics. 2021 April;btab227. doi:10.1093/bioinformatics/btab227
-
[25]
pyPESTO: a modular and scalable tool for parameter estimation for dynamic models
Sch¨ alte Y, Fr¨ ohlich F, Jost PJ, Vanhoefer J, Pathirana D, Stapor P, et al. pyPESTO: a modular and scalable tool for parameter estimation for dynamic models. Bioinformatics. 2023 11;39(11):btad711. doi:10.1093/bioinformatics/btad711
-
[26]
Fr¨ ohlich F, Sorger PK. Fides: Reliable trust-region optimization for parameter estimation of ordinary differential equation models. PLoS computational biology. 2022;18(7):e1010322. doi:10.1371/journal.pcbi.1010322
-
[27]
Penas DR, Gonz´ alez P, Egea JA, Doallo R, Banga JR. Parameter estimation in large-scale systems biology models: a parallel and self-adaptive cooperative strategy. BMC bioinformatics. 2017;18(1):52. doi:10.1186/s12859-016-1452-4
-
[28]
Triglyceride cycling enables modification of stored fatty acids
Wunderling K, Zurkovic J, Zink F, Kuerschner L, Thiele C. Triglyceride cycling enables modification of stored fatty acids. Nature Metabolism. 2023;5(4):699-709. doi:10.1038/s42255-023-00769-z
-
[29]
Benchmark problems for dynamic modeling of intracellular processes
Hass H, Loos C, Raim´ undez-´Alvarez E, Timmer J, Hasenauer J, Kreutz C. Benchmark problems for dynamic modeling of intracellular processes. Bioinformatics. 2019 01;35(17):3073-82. doi:10.1093/bioinformatics/btz020
-
[30]
Guidelines for benchmarking of optimization-based approaches for fitting mathematical models
Kreutz C. Guidelines for benchmarking of optimization-based approaches for fitting mathematical models. Genome Biology. 2019;20(1):281. doi:10.1186/s13059-019-1887-9
-
[31]
Benchmarking optimization methods for parameter estimation in large kinetic models
Villaverde AF, Froehlich F, Weindl D, Hasenauer J, Banga JR. Benchmarking optimization methods for parameter estimation in large kinetic models. Bioinformatics. 2018:bty736. doi:10.1093/bioinformatics/bty736
-
[32]
N¨ oh K, Niedenf¨ uhr S, Beyß M, Wiechert W. A Pareto approach to resolve the conflict between information gain and experimental costs: Multiple-criteria design of carbon labeling experiments. PLOS Computational Biology. 2018 10;14(10):e1006533. doi:10.1371/journal.pcbi.1006533
-
[33]
A survey on pareto front learning for multi-objective optimization
Kang S, Li K, Wang R. A survey on pareto front learning for multi-objective optimization. Journal of Membrane Computing. 2025;7(2):128-34. doi:10.1007/s41965-024-00170-z. June 12, 2026 21/21
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.