AF_Cache: Efficient Pipeline for Running AlphaFold for High-Throughput Protein-Protein Interaction Prediction
Pith reviewed 2026-06-28 03:19 UTC · model grok-4.3
The pith
AF_Cache pipeline reduces AlphaFold2 runtime for protein pairs by half and MSA generation by up to 13 times via caching and bucketing.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
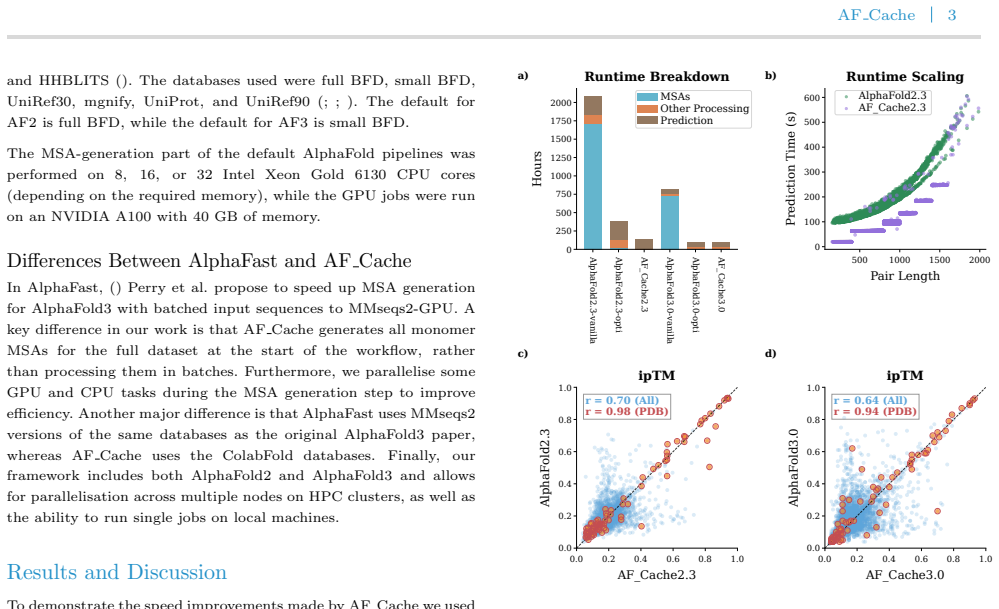
AF_Cache is a high-throughput pipeline that integrates GPU-accelerated MSA generation with MMseqs2, feature caching to eliminate redundant alignment computations, and sequence length bucketing to minimise repeated JAX compilations, yielding a ~2-fold reduction in AlphaFold2 inference time and up to a 13-fold speedup of MSA generation on 5,050 human mitochondrial protein pairs.
What carries the argument
Feature caching combined with sequence length bucketing inside a Nextflow workflow that calls MMseqs2 for MSA.
If this is right
- Large-scale screening of thousands of protein pairs becomes practical on modest hardware.
- The same caching and bucketing steps apply to both AlphaFold2 and AlphaFold3 workflows.
- Researchers gain a reusable Nextflow template for deploying structure-based PPI prediction at scale.
- Redundant MSA generation across overlapping pairs is avoided without altering the final models.
Where Pith is reading between the lines
- The same caching pattern could reduce cost in other alignment-heavy bioinformatics pipelines.
- If accuracy holds, the approach would support proteome-wide interaction maps that are currently too expensive.
- Speed gains may allow tighter integration with downstream tools such as docking or dynamics simulations.
Load-bearing premise
Caching alignments and bucketing sequences by length leave the AlphaFold interaction scores and structures unchanged.
What would settle it
Direct comparison of interaction scores or predicted structures between standard AlphaFold runs and AF_Cache runs on the same 5,050 mitochondrial pairs that reveals any consistent difference.
Figures

read the original abstract
Motivation: Accurate prediction of protein-protein interactions is essential for understanding biological processes, and recent advances such as AlphaFold2 and AlphaFold3 have enabled structure-based interaction prediction at unprecedented accuracy. However, the high computational cost of these methods, driven primarily by CPU-based repeated multiple sequence alignment (MSA) generation and, for AlphaFold2, repeated model recompilations, limits their applicability in large-scale, high-throughput settings. This creates a need for efficient pipelines that retain predictive performance while substantially reducing runtime. Results: We present AF_Cache, a high-throughput Nextflow pipeline for accelerating protein-protein interaction prediction using AlphaFold2 and AlphaFold3. AF_Cache combines GPU-accelerated MSA generation with MMseqs2, feature caching to eliminate redundant alignment computations, and sequence length bucketing to minimise repeated JAX compilations. Benchmarking on a dataset of 5,050 human mitochondrial protein pairs demonstrates a $\sim$2-fold reduction in inference time for AlphaFold2 and up to a 13-fold speedup of the MSA generation. AF\_Cache enables efficient large-scale interaction screening and provides a practical framework for deploying AlphaFold-based methods in high-throughput applications. Availability and implementation: The code and Nextflow pipeline are available on GitHub here: https://github.com/clami66/AF_cache. The code for reproducing the results of the paper, the MSAs, and the predicted models can be found at Zenodo: https://zenodo.org/records/20478892
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents AF_Cache, a Nextflow pipeline for high-throughput protein-protein interaction prediction with AlphaFold2 and AlphaFold3. It combines GPU-accelerated MSA generation using MMseqs2, feature caching to eliminate redundant computations, and sequence length bucketing to reduce JAX recompilations. Benchmarking on 5,050 human mitochondrial protein pairs reports a ~2-fold reduction in AlphaFold2 inference time and up to 13-fold speedup in MSA generation, with the claim that predictive performance is retained. The pipeline, reproduction code, MSAs, and models are publicly available on GitHub and Zenodo.
Significance. If the optimizations preserve original AlphaFold outputs, the work supplies a practical, reproducible framework for large-scale PPI screening that directly targets the dominant CPU and compilation bottlenecks. The explicit release of code, data, and models is a clear strength that enables independent verification and adoption.
major comments (1)
- [Abstract] Abstract (Results paragraph): the claim that AF_Cache 'retain[s] predictive performance' is unsupported by any reported metrics. No ipTM, pLDDT, RMSD, DockQ, or other equivalence statistics are provided comparing the baseline AlphaFold pipeline against the feature-cached and length-bucketed versions on the 5,050-pair set. Because feature caching assumes bit-identical MMseqs2 outputs and bucketing can alter padding/compilation paths, this verification is load-bearing for the central claim that the measured speedups are usable without accuracy loss.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on the need for explicit verification of retained predictive performance. We agree this is a substantive point and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Abstract] Abstract (Results paragraph): the claim that AF_Cache 'retain[s] predictive performance' is unsupported by any reported metrics. No ipTM, pLDDT, RMSD, DockQ, or other equivalence statistics are provided comparing the baseline AlphaFold pipeline against the feature-cached and length-bucketed versions on the 5,050-pair set. Because feature caching assumes bit-identical MMseqs2 outputs and bucketing can alter padding/compilation paths, this verification is load-bearing for the central claim that the measured speedups are usable without accuracy loss.
Authors: We agree that the claim requires supporting metrics and that the assumptions around bit-identical caching and bucketing do not substitute for empirical verification. In the revised manuscript we will add a dedicated results subsection (and associated supplementary table) that reports ipTM, pLDDT, and, where relevant, DockQ or RMSD values on a representative random subset of the 5,050 pairs, directly comparing the baseline AlphaFold2/3 pipeline against the cached and bucketed versions. The abstract will be updated to reference these equivalence results. We will also make the comparison scripts available in the reproduction repository. revision: yes
Circularity Check
No circularity; empirical runtime claims rest on direct measurements
full rationale
The paper is an engineering description of a Nextflow pipeline implementing feature caching and length bucketing for AlphaFold. All reported results are direct wall-clock timings on a fixed 5,050-pair mitochondrial dataset; no equations, fitted parameters, predictions derived from first principles, or uniqueness theorems appear. The preservation-of-accuracy premise is an unverified assumption (correctness issue) but is not part of any derivation chain that reduces to the inputs by construction. No self-citations are load-bearing for any mathematical claim.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Abagyan, R. A. and Batalov, S. , title =. J. Mol. Biol. , year = 1997, volume = 273, pages =
1997
-
[2]
and Taylor, W.R
Jones, D.T. and Taylor, W.R. and Thornton, J.M. A new appoach to protein fold recognition. Nature
-
[3]
and Schneider, R
Sander, C. and Schneider, R. Database of homology derived protein structures and the structural meaning of sequence alignment. Proteins: Struct. Funct. Genet
-
[4]
and Karplus, K
Park, J. and Karplus, K. and Barrett, C. and Hughey, R. and Haussler, D. and Hubbard, T. and Chothia C. , title =. J. Mol. Biol. , year = 1998, volume = 284, pages =
1998
-
[5]
and Eddy, S.R
Sonnhammer, E.L. and Eddy, S.R. and Durbin, R. Pfam: a Comprehensive database of protein domain families based on seed alignments. Proteins, Structure function and genetics. 1997
1997
-
[6]
and Teichmann, S
Park, J. and Teichmann, S. A. and Hubbard, T. and Chothia, C. , title =. J. Mol. Biol. , year = 1997, volume = 273, pages =
1997
-
[7]
and Apweiler, R
Bairoch, A. and Apweiler, R. The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Res
-
[8]
and Brenner, S.E
Murzin, A.G. and Brenner, S.E. and Hubbard, T. and Chothia, C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol
-
[9]
and Chothia, C
Lesk, A.M. and Chothia, C. How different amino acid sequences determine similar protein structures: the structure and evolutionary dynamics of the globins. J. Mol. Biol
-
[10]
and Lesk, A.M
Chothia, C. and Lesk, A.M. The relationship between the divergence of sequence and structure in proteins. EMBO J
-
[11]
and Madden, T.L
Altschul, S.F. and Madden, T.L. and Schaffer, A.A. and Zhang, J. and Zhang, Z. and Miller, W. and Lipman, D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997
1997
-
[12]
Brenner, S. E. and Chothia, C. and Hubbard, T. Assessing sequence comparison methods with reliable structurally identified evolutionary relationships. Proc. Natl. Acad. Sci. USA
-
[13]
Pearson, W. R. and Lipman, D. J. Improved Tools for Biological Sequence Analysis. Proc. Natl. Acad. Sci. U.S.A
-
[14]
Pearson, W. R. Comparison of methods for searching protein sequence databases. Protein Sci
-
[15]
and Sander, C
Holm, L. and Sander, C. Touring protein fold space with Dali/FSSP. Nucl. Acid. Res
-
[16]
and Waterman, M.S
Smith, T.F. and Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol
-
[17]
and Wunsch, C.D
Needleman, S.B. and Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol
-
[18]
and Henikoff, J.G
Henikoff, S. and Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA
-
[19]
and Cohen, M.A
Gonnet, G.H. and Cohen, M.A. and Benner, S.A. Exhaustive matching of the entire protein sequence database. Science
-
[20]
and Schwartz, R.M
Dayhoff, M. and Schwartz, R.M. and Orcutt, B.C. Atlas of protein sequence and structure
-
[21]
and Blaisdell, B
Karlin, S. and Blaisdell, B. E. and Mocarski, E. S. and Brendel, V. A method to identify distinctive charge configurations in protein sequences, with application to human herpesvirus polypeptides. J. Mol. Biol
-
[22]
Thompson, J. D. and Higgins, D.G and Gibson, T.J. , title =. Nucleic Acids Research , year = 1994, volume = 22, pages =
1994
-
[23]
and Sander, C
Rost, B. and Sander, C. Prediction of protein secondary structure structure at better than 70\. J. Mol. Biol
-
[24]
and Eisenberg, D
Rice, D. and Eisenberg, D. A 3 D -1 D substitution matrix for protein fold recognition that includes predicted secondary structure of the sequence. J. Mol. Biol
-
[25]
and Eisenberg, D
Fischer, D. and Eisenberg, D. Protein fold recognition using sequence-derived predictions. Protein Sci
-
[26]
and Schneider, R
Rost, B. and Schneider, R. and Sander, C. Protein fold recognition by prediction-based threading. J. Mol. Biol. 1997
1997
-
[27]
and Fischer, D
Rice, D. and Fischer, D. and Weiss, R. and Eisenberg, D. Fold assignments for amino acid sequences of the C A S P 2 experiment. Proteins: Struct. Funct. Genet., Suppl
-
[28]
and Geetha, V
Di Francesco, V. and Geetha, V. and Garnier, J. and Munson, P. J. Fold recognition using predicted secondary structure sequences and hidden M arkov models of proteins folds. Proteins: Struct. Funct. Genet., Suppl
-
[29]
and Domingues, F
Fl \"o ckner, H. and Domingues, F. and Sippl, M. J. Proteins folds from pair interactions: A blind test in fold recogition. Proteins: Struct. Funct. Genet., Suppl
-
[30]
and Sj \"o lander, K
Karplus, K. and Sj \"o lander, K. and Barrett, C. and Cline, M. and Haussler, D. and Hughey, R. and Holm, L. and Sander, C. Predicting structures using hidden M arkov models. Proteins: Struct. Funct. Genet., Suppl
-
[31]
and Lüthy, R
Bowie, J.U. and Lüthy, R. and Eisenberg, D. A method to identify protein sequence that fold into a known three-dimensional structure. Science
-
[32]
HMMER--Hidden M arkov model software URL: http://genome.wustl.edu/eddy/hmmer.html
Eddy, S.R. HMMER--Hidden M arkov model software URL: http://genome.wustl.edu/eddy/hmmer.html
-
[33]
and Eddy, S
Durbin, R. and Eddy, S. and Krogh, A. and Mitchison, G. Biological sequence analysis
-
[34]
and Brown, M
Krogh, A. and Brown, M. and Mian, I. S. and Sj \"o lander, K. and Haussler, D. Hidden M arkov models in computational biology: applications to protein modeling. J. Mol. Biol
-
[35]
and Hubbard, T
Moult, J. and Hubbard, T. and Bryant, S. H. and Fidelis, K. and Pedersen, J. T. Critical assesment of methods of proteins structure predictions ( C A S P ): Round I I. Proteins: Struct. Funct. Genet., Suppl
-
[36]
and Argos, P
Frishman, D. and Argos, P. Seventy-five percent accuracy in protein secondary structure prediction. Proteins: Struct. Funct. Genet
-
[37]
and Krogh, A
Haussler, D. and Krogh, A. and Mian, I. S. and Sj \"o lander, K. Protein modeling using hidden M arkov models: Analysis of globins. `` Hawaii International Conference on Systems Science ``
-
[38]
o lander, K. Protein modeling using hidden Markov models: Analysis of globins. \
Haussler, D. and Krogh, A. and Mian, I. S. and Sj \"o lander, K. Protein modeling using hidden Markov models: Analysis of globins. \" Hawaii International Conference on Systems Science
-
[39]
Rabiner, L. R. and Juang, B. H. An introduction to hidden M arkov models
-
[40]
Dayhoff, M. O. and Barker, W. C. and Hunt, L. T. Establishing homologies in protein sequences. Meth. Enzymol
-
[41]
Hubbard, T. J. and Park, J. , title =. Proteins: Struct. Funct. Genet. , year = 1995, volume = 23, pages =
1995
-
[42]
Fischer , title =
D. Fischer , title =. Pacific Symposium on Biocomputing , pages =
-
[43]
, title =
Krogh, A. , title =. Proc. of Fifth Int. Conf. on Intelligent Systems for Molecular Biology , pages =
-
[44]
and von Heijne, G
Sonnhammer, E. and von Heijne, G. and Krogh, A. , title =. Proc. of Sixth Int. Conf. on Intelligent Systems for Molecular Biology. , pages =
-
[45]
and Fischer, D
Elofsson, A. and Fischer, D. and Rice, D. W. and Le Grand, S. M. and Eisenberg D. , title =. Fold Des , year = 1996, volume = 1, pages =
1996
-
[46]
and McLachlan, A
Gribskov, M. and McLachlan, A. D. and Eisenberg, D. , title =. Proc Natl Acad Sci U S A , year = 1987, volume = 84, pages =
1987
-
[47]
and Levitt, M
Gerstein, M. and Levitt, M. , title =. Protein Sci , year = 1998, volume = 7, pages =
1998
-
[48]
and Sander, C
Holm, L. and Sander, C. , title =. Proteins: Struct. Funct. Genet. , year = 1997, volume = 28, pages =
1997
-
[49]
and Barrett, C
Karplus, K. and Barrett, C. and Hughey, R. , title =. Bioinformatics , year = 1998, volume = 14, pages =
1998
-
[50]
and Michi, A.D
Orengo, C.A. and Michi, A.D. and Jones, S. and Jones, D.T. and Swindels, M. B. and Thornton, J.M. , title =. Structure , year = 1997, volume = 5, pages =
1997
-
[51]
and Suwa, M
Salamov, A.A. and Suwa, M. and Orengo, C. A. and Swindells, M. B. , title =. Protein Sci , year = 1999, volume = 8, pages =
1999
-
[52]
Sheridan, R. P. and Dixon, J. S. and Venkataraghavan, R. , title =. Int. J. Pept. Protein Res. , year = 1985, volume = 25, pages =
1985
-
[53]
Salamov, A. A. and Suwa, M. and Orengo, C. A. and Swindells M. B. , title =. Protein Eng , year = 1999, volume = 12, pages =
1999
-
[54]
Lindahl, E and Elofsson, A , title =. J. Mol. Biol. , year = 2000, volume = 295, pages =
2000
-
[55]
Proc Natl Acad Sci U S A , year = 1998, volume = 95, number = 11, pages =
Levitt, M and Gerstein, M , title =. Proc Natl Acad Sci U S A , year = 1998, volume = 95, number = 11, pages =
1998
-
[56]
Proc Natl Acad Sci U S A , year = 1984, volume = 81, number = 4, pages =
Kabsch, W and Sander, C , title =. Proc Natl Acad Sci U S A , year = 1984, volume = 81, number = 4, pages =
1984
-
[57]
Fischer and A
D. Fischer and A. Elofsson and L. Rychlewski and F. Pazos and A. Valencia and B. Rost and Ortiz, A.R. and Dunbrack, R.L. , title =. Proteins , year =
-
[58]
1999 , title =
CASP , key =. 1999 , title =
1999
-
[59]
and Barret, C
Fischer, D. and Barret, C. and Bryson, K. and Elofsson, A. and Godzik, A. and Jones, D. and Karplus, K.J. and Kelley, L.A. and MacCallum, R.M. and Pawowski, K. and Rost, B. and Rychlewski, L. and Sternberg, M. , title =. Proteins , year =
-
[60]
Siew and A
N. Siew and A. Elofsson and L. Rychlewski and D. Fischer , title =. Bionformatics , year =
-
[61]
Jones and G.J
T.A. Jones and G.J. Kleywegt , title =. Proteins , year = 1999, volume =
1999
-
[62]
Orengo and J.E
C.A. Orengo and J.E. Bray and T. Hubbard and L. LoConte and I. Sillitoe , title =. Proteins , year = 1999, volume =
1999
-
[63]
Zemla and C
A. Zemla and C. Veclovas and J. Moult and K. Fidelis , title =. Proteins , year = 1999, volume =
1999
-
[64]
Proteins , year = 1999, volume =
T.J.P Hubbard , title =. Proteins , year = 1999, volume =
1999
-
[65]
Murzin , title =
A.G. Murzin , title =. Proteins , year = 1999, volume =
1999
-
[66]
Moult and T
J. Moult and T. Hubbard and K. Fidelis and J.T. Pedersen , title =. Proteins , year = 1999, volume =
1999
-
[67]
Feng and M.J
Z.-K. Feng and M.J. Sippl , title =. Fold. Des. , year = 1996, volume = 1, pages =
1996
-
[68]
Abagyan and M.M
R.A. Abagyan and M.M. Totrov , title =. J. Mol. Biol , year = 1997, volume = 268, number = 3, pages =
1997
-
[69]
and Persson, B
Kallberg, Y. and Persson, B. , title =. Bioinformatics , year = 1999, volume = 15, number = 3, pages =
1999
-
[70]
Rost , title =
B. Rost , title =. Proteins , year = 1997, volume =
1997
-
[71]
, title =
Sali, A. , title =. Nat Struct Biol , year = 1998, volume = 5, number = 12, pages =
1998
-
[72]
, title =
Sanchez, R and Sali, A. , title =
-
[73]
Peitsch and M.R
M.C. Peitsch and M.R. Wilkins and L. Tonella and J-C Sanchez and D.F Hochstrasser , title =. Electrophoresis , year = 1997, volume = 18, pages =
1997
-
[74]
Jones , title =
D.T. Jones , title =
-
[75]
, title =
Jones, D.T. , title =
-
[76]
Alexandrov and R
N. Alexandrov and R. Lüthy , title =. Protein Sci. , year = 1998, volume = 7, pages =
1998
-
[77]
and Bowie, J.U
Lüthy, R. and Bowie, J.U. and Eisenberg, D. , title =. Nature , year = 1992, volume = 356, number = 6364, pages =
1992
-
[78]
Domingues and P
F.S. Domingues and P. Lackner and A. Andreeva and M.J. Sippl , title =. J. Mol. Biol , year = 2000, volume = 297, number = 4, pages =
2000
-
[79]
Rost , title =
B. Rost , title =. Protein Eng. , year = 1999, volume = 12, number = 2, pages =
1999
-
[80]
Hadley and D
C. Hadley and D. T. Jones , title =. Structure , year = 1999, volume = 7, number = 8, pages =
1999
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.