Recognition: 1 theorem link
· Lean TheoremResource Estimation for VQE on Small Molecules: Impact of Fermion Mappings and Hamiltonian Reductions
Pith reviewed 2026-05-17 03:00 UTC · model grok-4.3
The pith
Suitable fermion mappings combined with symmetry reductions can halve qubit counts and cut gate counts by up to 27.5 times for VQE calculations on small molecules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The analysis reveals that appropriate transformations, when combined with symmetry-based reductions, can substantially reduce qubit counts by up to ≈50% and quantum gate counts by up to ≈27.5× and Hamiltonian Pauli string counts by up to ≈2.75×, relative to the corresponding unreduced Hamiltonian representations within the same active-space configuration for the representative set of molecular systems under study.
What carries the argument
The Jordan-Wigner, Bravyi-Kitaev, and Parity fermion-to-qubit mappings together with Z2 tapering and frozen-core approximations, which shrink the effective qubit register size and remove redundant terms from the qubit Hamiltonian.
If this is right
- VQE circuits for the tested molecules fit on smaller NISQ processors than the unreduced versions would allow.
- The measured resource numbers supply concrete baselines for estimating costs when moving the same molecules to fault-tolerant architectures.
- Circuit designers can prioritize specific mappings and tapering steps to minimize depth and term count for a given active space.
- The same reduction pipeline applies directly to other small molecules without altering the underlying active-space choice.
Where Pith is reading between the lines
- The reported savings might change if a different ansatz, such as a hardware-efficient one, replaces UCCSD.
- Combining these reductions with error-mitigation techniques could further improve effective accuracy on noisy hardware.
- Testing the same mappings and reductions on slightly larger active spaces would show how the resource gains scale with molecular size.
Load-bearing premise
The UCCSD ansatz together with the chosen active spaces and reductions must still produce ground-state energies accurate to chemical precision without the simplifications adding errors that would invalidate the reported resource counts.
What would settle it
For a concrete molecule such as LiH or H2O, run the reduced VQE circuit, compare its ground-state energy to a high-accuracy classical reference, and check whether the energy error remains below 1.6 milliHartree while the measured qubit, gate, and Pauli-term counts match the stated reduction factors.
Figures

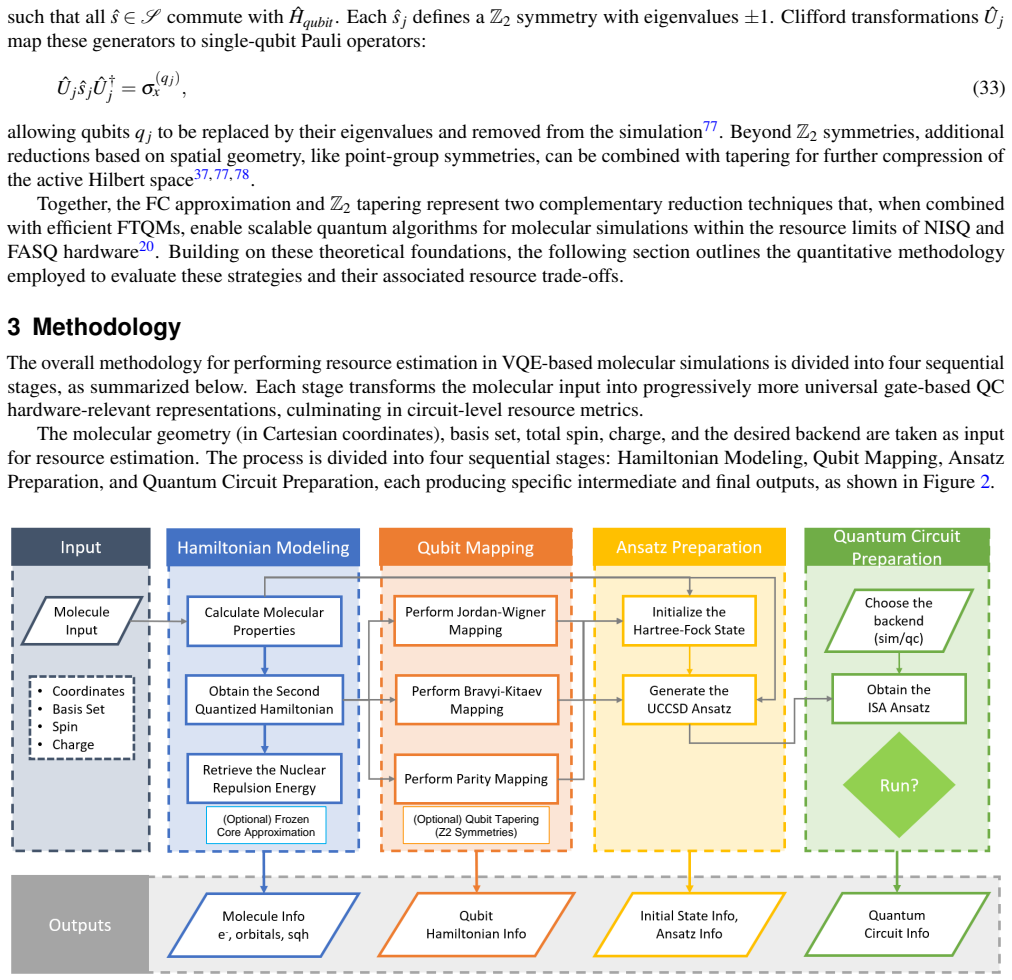



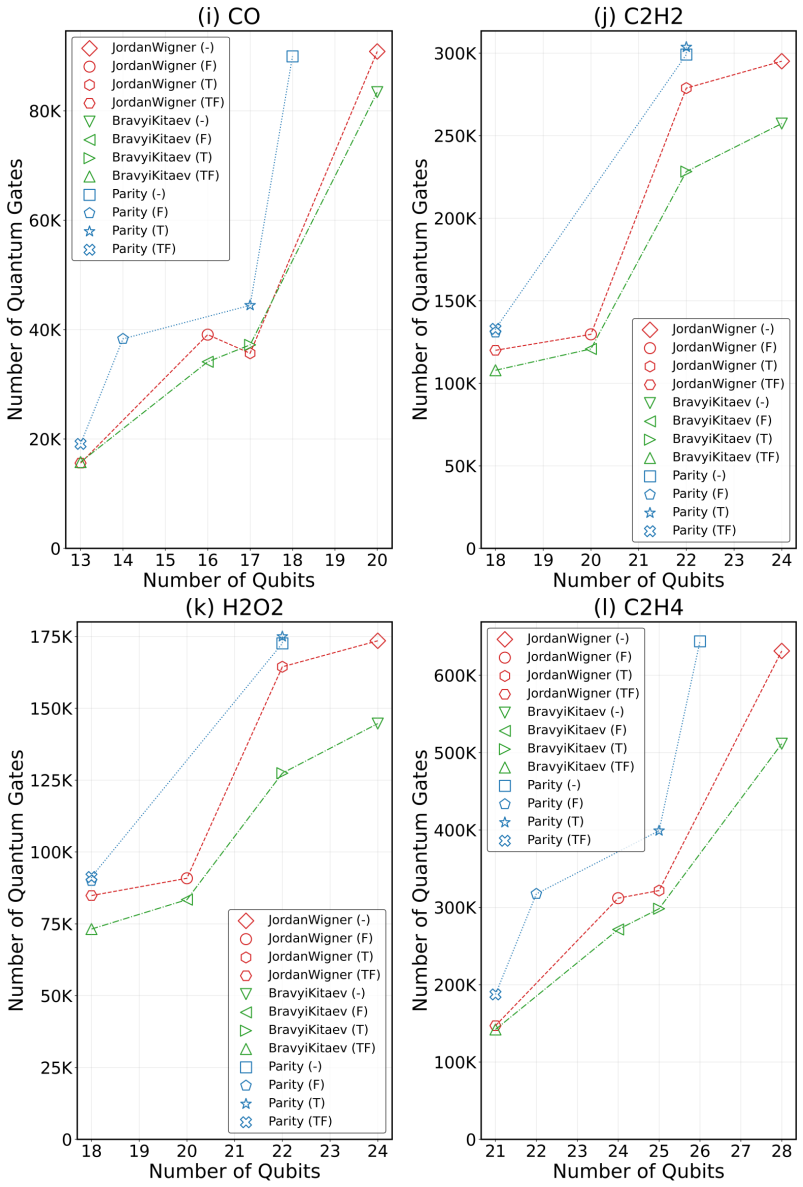
read the original abstract
Accurate determination of ground-state energies for molecules remains a challenge in quantum chemistry and a cornerstone for progress in fields such as drug discovery and materials design. The Variational Quantum Eigensolver (VQE) represents a leading hybrid quantum-classical paradigm for addressing this challenge; however, its widespread realization is limited by noise and the restricted scalability of current quantum hardware. Achieving efficient simulations on Noisy Intermediate-Scale Quantum (NISQ) devices and forthcoming Fault-Tolerant Application-Scalable Quantum (FASQ) systems demands a detailed understanding of how computational resources scale with molecular complexity and fermion-to-qubit encodings. In this study, resource requirements for VQE implementations employing the Unitary Coupled Cluster Singles and Doubles (UCCSD) ansatz are systematically analyzed. The molecular Hamiltonian is formulated in second quantization and mapped to qubit operators through the Jordan-Wigner (JW), Bravyi-Kitaev (BK), and Parity (Pa) transformations. Hamiltonian reduction strategies, including $\mathbb{Z}_2$ tapering and frozen-core approximations, are examined to assess their effect on quantum resource scaling. The analysis reveals that appropriate transformations, when combined with symmetry-based reductions, can substantially reduce qubit counts by up to $\approx 50\%$ and quantum gate counts by up to $\approx 27.5\times$ and Hamiltonian Pauli string counts by up to $\approx 2.75\times$, relative to the corresponding unreduced Hamiltonian representations within the same active-space configuration for the representative set of molecular systems under study. These findings provide practical circuit-level insights for executing chemically relevant simulations on NISQ hardware, while establishing physical-resource baselines that may inform future logical-level analyses targeting FASQ systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper analyzes resource requirements for VQE with the UCCSD ansatz on small molecules. It compares Jordan-Wigner, Bravyi-Kitaev, and Parity fermion-to-qubit mappings, then applies Z2 tapering and frozen-core reductions to the second-quantized Hamiltonian. The central claim is that these steps, within the same active-space configuration, yield up to ~50% fewer qubits, ~27.5× fewer quantum gates, and ~2.75× fewer Pauli strings relative to the unreduced active-space Hamiltonian for the studied molecules, providing practical baselines for NISQ and FASQ implementations.
Significance. If the resource counts are shown to apply to chemically accurate ground states, the work supplies concrete, mapping-specific scaling data derived from explicit circuit constructions. This is useful for NISQ circuit design and for setting logical-resource targets on fault-tolerant hardware. The systematic side-by-side comparison of three mappings plus two reduction techniques is a clear strength.
major comments (2)
- [Abstract] Abstract: the claim that savings occur 'within the same active-space configuration' is undercut by the inclusion of frozen-core approximations. Unlike exact Z2 tapering, frozen-core discards core orbitals and correlation, shifting the ground-state energy by an amount that depends on the molecule and basis and can exceed chemical accuracy (~1.6 mHa). The reported 50% qubit, 27.5× gate, and 2.75× Pauli-string reductions therefore apply to an approximated Hamiltonian rather than the target chemically accurate problem; the UCCSD ansatz does not correct this Hamiltonian-level error.
- [Resource Estimation] Resource Estimation section (or equivalent): the manuscript provides no quantitative verification that the frozen-core energies remain within chemical accuracy for the tabulated molecules, nor does it separate the resource savings attributable to exact Z2 tapering from those attributable to the approximate frozen-core step. Without these data the central scaling claims cannot be interpreted as applying to the intended problem.
minor comments (2)
- [Abstract] The abstract and introduction would benefit from an explicit list of the molecular systems, basis sets, and active-space sizes used in the study.
- [Methods] Clarify how gate counts were obtained after transpilation (e.g., which compiler, optimization level, and whether two-qubit gate counts or total gate counts are reported).
Simulated Author's Rebuttal
We thank the referee for their insightful comments and the opportunity to clarify aspects of our work. We respond to each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that savings occur 'within the same active-space configuration' is undercut by the inclusion of frozen-core approximations. Unlike exact Z2 tapering, frozen-core discards core orbitals and correlation, shifting the ground-state energy by an amount that depends on the molecule and basis and can exceed chemical accuracy (~1.6 mHa). The reported 50% qubit, 27.5× gate, and 2.75× Pauli-string reductions therefore apply to an approximated Hamiltonian rather than the target chemically accurate problem; the UCCSD ansatz does not correct this Hamiltonian-level error.
Authors: We thank the referee for highlighting this important distinction. The phrase 'within the same active-space configuration' was meant to convey that the fermion mappings and Z2 tapering are applied to Hamiltonians derived from the same selection of active orbitals. Frozen-core approximation is a standard method in quantum chemistry to reduce the problem size by freezing core electrons, which is part of defining the active space for many calculations. Nevertheless, we recognize that this step introduces an approximation to the energy. In the revised manuscript, we will update the abstract to more precisely describe the reductions as including frozen-core approximations and will add a sentence noting that the accuracy of the frozen-core approach for the studied systems is consistent with common practices in the field, where it often preserves chemical accuracy for valence-dominated properties. revision: yes
-
Referee: [Resource Estimation] Resource Estimation section (or equivalent): the manuscript provides no quantitative verification that the frozen-core energies remain within chemical accuracy for the tabulated molecules, nor does it separate the resource savings attributable to exact Z2 tapering from those attributable to the approximate frozen-core step. Without these data the central scaling claims cannot be interpreted as applying to the intended problem.
Authors: We agree that explicit verification and separation of effects would enhance the interpretability of our results. Although the current manuscript focuses on resource scaling rather than energy accuracy benchmarks, we will incorporate in the revision a table or discussion providing the frozen-core energy errors for the molecules considered, drawing from established quantum chemistry literature or our own calculations where possible. Furthermore, we will add resource estimates broken down by the application of Z2 tapering alone versus the combined Z2 tapering and frozen-core reductions to clearly separate the contributions of each technique. revision: yes
Circularity Check
Resource counts obtained via direct enumeration from mappings and reductions; no circular reduction to inputs.
full rationale
The paper derives qubit, gate, and Pauli-string counts through explicit application of JW/BK/Pa mappings, Z2 tapering, and frozen-core approximations to the second-quantized Hamiltonian, followed by UCCSD circuit construction. These are standard, non-fitted enumerations performed on concrete molecular instances within fixed active spaces. No equation or central claim reduces the reported savings (e.g., 50% qubit reduction) to a parameter defined by the same data or to a self-citation chain. The derivation chain remains self-contained against external benchmarks of Hamiltonian term counting and circuit depth estimation.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption UCCSD ansatz accurately captures the ground-state wavefunction for the studied molecules within the chosen active space
- domain assumption Frozen-core and Z2 tapering preserve the essential chemistry without significant accuracy loss
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Hamiltonian reduction strategies, including Z2 tapering and frozen-core approximations, are examined to assess their effect on quantum resource scaling.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Forward citations
Cited by 1 Pith paper
-
Loss-aware state space geometry for quantum variational algorithms
Loss-aware natural gradient variants are introduced by embedding the loss hypersurface in a statistical manifold or using quantum state overlaps, yielding conformal updates that adjust effective step size.
Reference graph
Works this paper leans on
-
[1]
Feynman, R. P. Simulating physics with computers.Int. J. Theor. Phys.21, 467–488, DOI: 10.1007/BF02650179 (1982)
-
[2]
Tonomura, A., Endo, J., Matsuda, T., Kawasaki, T. & Ezawa, H. Demonstration of single-electron buildup of an interference pattern.Am. J. Phys.57, 117–120, DOI: 10.1119/1.16104 (1989)
-
[3]
Experiment and the foundations of quantum physics.Rev
Zeilinger, A. Experiment and the foundations of quantum physics.Rev. Mod. Phys.71, S288–S297, DOI: 10.1103/ RevModPhys.71.S288 (1999)
work page 1999
-
[4]
Einstein, A., Podolsky, B. & Rosen, N. Can quantum-mechanical description of physical reality be considered complete? Phys. Rev.47, 777–780, DOI: 10.1103/PhysRev.47.777 (1935)
-
[5]
Horodecki, R., Horodecki, P., Horodecki, M. & Horodecki, K. Quantum entanglement.Rev. Mod. Phys.81, 865–942, DOI: 10.1103/RevModPhys.81.865 (2009)
-
[6]
Algorithms for quantum computation: discrete logarithms and factoring
Shor, P. Algorithms for quantum computation: discrete logarithms and factoring. InProceedings 35th Annual Symposium on F oundations of Computer Science, 124–134, DOI: 10.1109/SFCS.1994.365700 (1994)
-
[7]
Grover, L. K. A fast quantum mechanical algorithm for database search. InProceedings of the Twenty-Eighth Annual ACM Symposium on Theory of Computing, STOC ’96, 212–219, DOI: 10.1145/237814.237866 (Association for Computing Machinery, New York, NY , USA, 1996)
-
[8]
Shor, P. W. Polynomial-time algorithms for prime factorization and discrete logarithms on a quantum computer.SIAM J. on Comput.26, 1484–1509, DOI: 10.1137/S0097539795293172 (1997)
-
[9]
& Chuang, I.Quantum Computation and Quantum Information
Nielsen, M. & Chuang, I.Quantum Computation and Quantum Information. Cambridge Series on Information and the Natural Sciences (Cambridge University Press, 2000). 11.Lloyd, S. Universal quantum simulators.Science273, 1073–1078, DOI: 10.1126/science.273.5278.1073 (1996)
-
[10]
Aspuru-Guzik, A., Dutoi, A. D., Love, P. J. & Head-Gordon, M. Simulated quantum computation of molecular energies. Science309, 1704–1707, DOI: 10.1126/science.1113479 (2005)
-
[11]
A Quantum Approximate Optimization Algorithm
Harrow, A. W., Hassidim, A. & Lloyd, S. Quantum algorithm for linear systems of equations.Phys. Rev. Lett.103, 150502, DOI: 10.1103/PhysRevLett.103.150502 (2009). 14.Farhi, E., Goldstone, J. & Gutmann, S. A quantum approximate optimization algorithm (2014). 1411.4028
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1103/physrevlett.103.150502 2009
-
[12]
Boyer, M., Brassard, G., Høyer, P. & Tapp, A. Tight bounds on quantum searching.F ortschritte der Physik46, 493–505, DOI: 10.1002/(SICI)1521-3978(199806)46:4/5<493::AID-PROP493>3.0.CO;2-P (1998)
-
[13]
Rev.119, 10856–10915, DOI: 10.1021/acs
Cao, Y .et al.Quantum chemistry in the age of quantum computing.Chem. Rev.119, 10856–10915, DOI: 10.1021/acs. chemrev.8b00803 (2019)
work page doi:10.1021/acs 2019
-
[14]
McArdle, S., Endo, S., Aspuru-Guzik, A., Benjamin, S. C. & Yuan, X. Quantum computational chemistry.Rev. Mod. Phys.92, 015003, DOI: 10.1103/RevModPhys.92.015003 (2020)
-
[15]
Patra, A. K.et al.Survey of quantum algorithms: Foundations, frameworks and applications.AuthoreaDOI: 10.22541/ au.176341037.72812424/v1 (2025)
-
[16]
Bauer, B., Bravyi, S., Motta, M. & Chan, G. K.-L. Quantum algorithms for quantum chemistry and quantum materials science.Chem. Rev.120, 12685–12717, DOI: 10.1021/acs.chemrev.9b00829 (2020). 15/19
-
[17]
O’Malley, P. J. J.et al.Scalable quantum simulation of molecular energies.Phys. Rev. X6, 031007, DOI: 10.1103/ PhysRevX.6.031007 (2016)
work page 2016
-
[18]
Quantum Computing in the NISQ era and beyond.Quantum2, 79, DOI: 10.22331/q-2018-08-06-79 (2018)
Preskill, J. Quantum Computing in the NISQ era and beyond.Quantum2, 79, DOI: 10.22331/q-2018-08-06-79 (2018). 22.Eisert, J. & Preskill, J. Mind the gaps: The fraught road to quantum advantage (2025). 2510.19928
work page internal anchor Pith review doi:10.22331/q-2018-08-06-79 2018
-
[19]
Peruzzo, A.et al.A variational eigenvalue solver on a photonic quantum processor.Nat. Commun.5, 4213, DOI: 10.1038/ncomms5213 (2014)
-
[20]
McClean, J. R., Romero, J., Babbush, R. & Aspuru-Guzik, A. The theory of variational hybrid quantum-classical algorithms.New J. Phys.18, 023023, DOI: 10.1088/1367-2630/18/2/023023 (2016)
-
[21]
Barkoutsos, P. K.et al.Quantum algorithms for electronic structure calculations: Particle-hole hamiltonian and optimized wave-function expansions.Phys. Rev. A98, 022322, DOI: 10.1103/PhysRevA.98.022322 (2018)
-
[22]
Reports986, 1–128, DOI: 10.1016/j.physrep.2022.08.003 (2022)
Tilly, J.et al.The variational quantum eigensolver: A review of methods and best practices.Phys. Reports986, 1–128, DOI: 10.1016/j.physrep.2022.08.003 (2022). The Variational Quantum Eigensolver: a review of methods and best practices
-
[23]
Tranter, A., Love, P. J., Mintert, F. & Coveney, P. V . A comparison of the bravyi–kitaev and jordan–wigner transformations for the quantum simulation of quantum chemistry.J. Chem. Theory Comput.14, 5617–5630, DOI: 10.1021/acs.jctc. 8b00450 (2018)
-
[24]
Kühn, M., Zanker, S., Deglmann, P., Marthaler, M. & Weiß, H. Accuracy and resource estimations for quantum chemistry on a near-term quantum computer.J. Chem. Theory Comput.15, 4764–4780, DOI: 10.1021/acs.jctc.9b00236 (2019)
-
[25]
Resource estimation for quantum variational simulations of the hubbard model.Phys
Cai, Z. Resource estimation for quantum variational simulations of the hubbard model.Phys. Rev. Appl.14, 014059, DOI: 10.1103/PhysRevApplied.14.014059 (2020)
-
[26]
Johnson, P. D.et al.Reducing the cost of energy estimation in the variational quantum eigensolver algorithm with robust amplitude estimation (2022). 2203.07275
-
[27]
Otten, M.et al.Qrechem: quantum resource estimation software for chemistry applications.Front. Quantum Sci. Technol. 2, DOI: 10.3389/frqst.2023.1232624 (2023)
-
[28]
Patel, S., Jayakumar, P., Yen, T.-C. & Izmaylov, A. F. Quantum measurement for quantum chemistry on a quantum computer.Chem. Rev.125, 7490–7524, DOI: 10.1021/acs.chemrev.5c00055 (2025)
-
[29]
Belaloui, N. E.et al.Ground-state energy estimation on current quantum hardware through the variational quantum eigensolver: A practical study.J. Chem. Theory Comput.21, 6777–6792, DOI: 10.1021/acs.jctc.4c01657 (2025). 34.Gundlach, H.et al.Quantum advantage in computational chemistry? (2025). 2508.20972
-
[30]
Alexeev, Y .et al.A perspective on quantum computing applications in quantum chemistry using 25–100 logical qubits.J. Chem. Theory Comput.DOI: 10.1021/acs.jctc.5c01038 (2025)
-
[31]
Technol.4, 014008, DOI: 10.1088/2058-9565/aad3e4 (2018)
Romero, J.et al.Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz.Quantum Sci. Technol.4, 014008, DOI: 10.1088/2058-9565/aad3e4 (2018)
-
[32]
Tapering off qubits to simulate fermionic Hamiltonians
Bravyi, S., Gambetta, J. M., Mezzacapo, A. & Temme, K. Tapering off qubits to simulate fermionic hamiltonians (2017). 1701.08213
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[33]
Jordan, P. & Wigner, E. Über das paulische äquivalenzverbot.Zeitschrift für Physik47, 631–651, DOI: 10.1007/ BF01331938 (1928)
work page 1928
-
[34]
A.et al.The fermionic canonical commutation relations and the jordan-wigner transform.Sch
Nielsen, M. A.et al.The fermionic canonical commutation relations and the jordan-wigner transform.Sch. Phys. Sci. The Univ. Qld.59, 75 (2005)
work page 2005
-
[35]
Bravyi, S. B. & Kitaev, A. Y . Fermionic quantum computation.Annals Phys.298, 210–226, DOI: 10.1006/aphy.2002.6254 (2002)
-
[36]
Seeley, J. T., Richard, M. J. & Love, P. J. The bravyi-kitaev transformation for quantum computation of electronic structure.The J. Chem. Phys.137, 224109, DOI: 10.1063/1.4768229 (2012)
-
[37]
Tranter, A.et al.The bravyi–kitaev transformation: Properties and applications.Int. J. Quantum Chem.115, 1431–1441, DOI: 10.1002/qua.24969 (2015)
-
[38]
Szabo, A. & Ostlund, N. S.Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, chap. 2, 39–107. Dover Books on Chemistry (Dover Publications, Mineola, NY , 1996), 1st, revised edn. 16/19
work page 1996
-
[39]
& Olsen, J.Molecular Electronic-Structure Theory, chap
Helgaker, T., Jørgensen, P. & Olsen, J.Molecular Electronic-Structure Theory, chap. 1, 1–33 (John Wiley & Sons, Ltd, 2000), 1st edn. 45.Born, M. & Huang, K.Dynamical Theory of Crystal Lattices(Clarendon Press, Oxford, 1954)
work page 2000
-
[40]
Fetter, A. L. & Walecka, J. D.Quantum Theory of Many-Particle Systems, chap. 1, 3–32. Dover Books on Physics (Dover Publications, Mineola, New York, 2003). Corrected reprint of the 7th printing (McGraw-Hill, 1971)
work page 2003
-
[41]
Shavitt, I. & Bartlett, R. J.Many-Body Methods in Chemistry and Physics: MBPT and Coupled-Cluster Theory, chap. 2, 54–90. Cambridge Molecular Science (Cambridge University Press, Cambridge, 2009), 1st edn. Chapter 2. 48.Born, M. & Oppenheimer, R. Zur quantentheorie der molekeln.Annalen der Physik389, 457–484, DOI: 10.1002/andp. 19273892002 (1927)
-
[42]
Thomas, I. L. Protonic structure of molecules. i. ammonia molecules.Phys. Rev.185, 90–94, DOI: 10.1103/PhysRev.185. 90 (1969)
-
[43]
The protonic structure of methane, ammonia, water, and hydrogen fluoride.Chem
Thomas, I. The protonic structure of methane, ammonia, water, and hydrogen fluoride.Chem. Phys. Lett.3, 705–706, DOI: 10.1016/0009-2614(69)87015-6 (1969)
-
[44]
Tachikawa, M., Mori, K., Nakai, H. & Iguchi, K. An extension of ab initio molecular orbital theory to nuclear motion. Chem. Phys. Lett.290, 437–442, DOI: 10.1016/S0009-2614(98)00519-3 (1998)
-
[45]
Nakai, H., Sodeyama, K. & Hoshino, M. Non-born–oppenheimer theory for simultaneous determination of vibrational and electronic excited states: ab initio no+mo/cis theory.Chem. Phys. Lett.345, 118–124, DOI: 10.1016/S0009-2614(01) 00836-3 (2001)
-
[46]
Nakai, H. Simultaneous determination of nuclear and electronic wave functions without born–oppenheimer approximation: Ab initio no+mo/hf theory.Int. J. Quantum Chem.86, 511–517, DOI: 10.1002/qua.1106 (2002)
-
[47]
Nakai, H. & Sodeyama, K. Many-body effects in nonadiabatic molecular theory for simultaneous determination of nuclear and electronic wave functions: Ab initio nomo/mbpt and cc methods.The J. Chem. Phys.118, 1119–1127, DOI: 10.1063/1.1528951 (2003)
-
[48]
Nakai, H. Nuclear orbital plus molecular orbital theory: Simultaneous determination of nuclear and electronic wave functions without born–oppenheimer approximation.Int. J. Quantum Chem.107, 2849–2869, DOI: 10.1002/qua.21379 (2007)
-
[49]
Tachikawa, M. Simultaneous optimization of gaussian type function exponents for electron and positron with full-ci wavefunction – application to ground and excited states of positronic compounds with multi-component molecular orbital approach.Chem. Phys. Lett.350, 269–276, DOI: 10.1016/S0009-2614(01)01286-6 (2001)
-
[50]
Ishimoto, T., Tachikawa, M. & Nagashima, U. Review of multicomponent molecular orbital method for direct treatment of nuclear quantum effect.Int. J. Quantum Chem.109, 2677–2694, DOI: 10.1002/qua.22069 (2009)
-
[51]
Webb, S. P., Iordanov, T. & Hammes-Schiffer, S. Multiconfigurational nuclear-electronic orbital approach: Incorporation of nuclear quantum effects in electronic structure calculations.The J. Chem. Phys.117, 4106–4118, DOI: 10.1063/1.1494980 (2002)
-
[52]
Pavoševi´c, F., Culpitt, T. & Hammes-Schiffer, S. Multicomponent quantum chemistry: Integrating electronic and nuclear quantum effects via the nuclear–electronic orbital method.Chem. Rev.120, 4222–4253, DOI: 10.1021/acs.chemrev. 9b00798 (2020)
-
[53]
Xu, X. & Yang, Y . Constrained nuclear-electronic orbital density functional theory: Energy surfaces with nuclear quantum effects.The J. Chem. Phys.152, 084107, DOI: 10.1063/1.5143371 (2020)
-
[54]
Xu, X. & Yang, Y . Full-quantum descriptions of molecular systems from constrained nuclear–electronic orbital density functional theory.The J. Chem. Phys.153, 074106, DOI: 10.1063/5.0014001 (2020)
-
[55]
Culpitt, T., Chen, Z., Pavoševi´c, F. & Yang, Y . Constrained nuclear-electronic orbital theory for quantum computation.J. Chem. Theory Comput.21, 7845–7854, DOI: 10.1021/acs.jctc.5c00815 (2025)
-
[56]
Hehre, W. J., Stewart, R. F. & Pople, J. A. Self-consistent molecular-orbital methods. i. use of gaussian expansions of slater-type atomic orbitals.The J. Chem. Phys.51, 2657–2664, DOI: 10.1063/1.1672392 (1969)
-
[57]
Gaussian basis sets for use in correlated molecular calculations
Dunning, J., Thom H. Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen.The J. Chem. Phys.90, 1007–1023, DOI: 10.1063/1.456153 (1989)
-
[58]
Fock, V . Konfigurationsraum und zweite quantelung.Zeitschrift für Physik75, 622–647, DOI: 10.1007/BF01344458 (1932). 17/19
-
[59]
Steudtner, M. & Wehner, S. Fermion-to-qubit mappings with varying resource requirements for quantum simulation. New J. Phys.20, 063010, DOI: 10.1088/1367-2630/aac54f (2018)
-
[60]
Jiang, Z., Kalev, A., Mruczkiewicz, W. & Neven, H. Optimal fermion-to-qubit mapping via ternary trees with applications to reduced quantum states learning.Quantum4, 276, DOI: 10.22331/q-2020-06-04-276 (2020)
-
[61]
Derby, C., Klassen, J., Bausch, J. & Cubitt, T. Compact fermion to qubit mappings.Phys. Rev. B104, 035118, DOI: 10.1103/PhysRevB.104.035118 (2021)
-
[62]
Miller, A., Zimborás, Z., Knecht, S., Maniscalco, S. & García-Pérez, G. Bonsai algorithm: Grow your own fermion-to- qubit mappings.PRX Quantum4, 030314, DOI: 10.1103/PRXQuantum.4.030314 (2023)
-
[63]
O’Brien, O. & Strelchuk, S. Ultrafast hybrid fermion-to-qubit mapping.Phys. Rev. B109, 115149, DOI: 10.1103/ PhysRevB.109.115149 (2024)
work page 2024
-
[64]
72.Yu, J., Liu, Y ., Sugiura, S., Van V oorhis, T
Parella-Dilmé, T.et al.Reducing entanglement with physically inspired fermion-to-qubit mappings.PRX Quantum5, 030333, DOI: 10.1103/PRXQuantum.5.030333 (2024). 72.Yu, J., Liu, Y ., Sugiura, S., Van V oorhis, T. & Zeytino˘glu, S. Clifford circuit-based heuristic optimization of fermion-to- qubit mappings.J. Chem. Theory Comput.21, 9430–9443, DOI: 10.1021/ac...
-
[65]
Liu, Y .et al.Hatt: Hamiltonian adaptive ternary tree for optimizing fermion-to-qubit mapping. In2025 IEEE International Symposium on High Performance Computer Architecture (HPCA), 143–157, DOI: 10.1109/HPCA61900.2025.00022 (2025)
-
[66]
Roos, B. O., Taylor, P. R. & Sigbahn, P. E. A complete active space scf method (casscf) using a density matrix formulated super-ci approach.Chem. Phys.48, 157–173, DOI: 10.1016/0301-0104(80)80045-0 (1980)
-
[67]
Battaglia, S., Rossmannek, M., Rybkin, V . V ., Tavernelli, I. & Hutter, J. A general framework for active space embedding methods with applications in quantum computing.npj Comput. Mater.10, 297, DOI: 10.1038/s41524-024-01477-2 (2024)
-
[68]
Gard, B. T.et al.Efficient symmetry-preserving state preparation circuits for the variational quantum eigensolver algorithm.npj Quantum Inf.6, 10, DOI: 10.1038/s41534-019-0240-1 (2020)
-
[69]
Setia, K.et al.Reducing qubit requirements for quantum simulations using molecular point group symmetries.J. Chem. Theory Comput.16, 6091–6097, DOI: 10.1021/acs.jctc.0c00113 (2020)
-
[70]
Seki, K., Shirakawa, T. & Yunoki, S. Symmetry-adapted variational quantum eigensolver.Phys. Rev. A101, 052340, DOI: 10.1103/PhysRevA.101.052340 (2020)
-
[71]
Fock, V . Näherungsmethode zur lösung des quantenmechanischen mehrkörperproblems.Zeitschrift für Physik61, 126–148, DOI: 10.1007/BF01340294 (1930). 80.Van Rossum, G. Python tutorial. Tech. Rep. CS-R9526, Centrum voor Wiskunde en Informatica (CWI) (1995). 81.Python Software Foundation.Python Language Reference, v3.11(2022). Accessed: 2025-11-18. 82.Harris,...
-
[72]
Libcint: An efficient general integral library for gaussian basis functions.J
Sun, Q. Libcint: An efficient general integral library for gaussian basis functions.J. Comput. Chem.36, 1664–1671, DOI: https://doi.org/10.1002/jcc.23981 (2015)
-
[73]
Sun, Q.et al.Pyscf: the python-based simulations of chemistry framework.WIREs Comput. Mol. Sci.8, e1340, DOI: https://doi.org/10.1002/wcms.1340 (2018)
-
[74]
Sun, Q.et al.Recent developments in the pyscf program package.The J. Chem. Phys.153, 024109, DOI: 10.1063/5. 0006074 (2020). 86.Javadi-Abhari, A.et al.Quantum computing with qiskit (2024). 2405.08810. 87.Qiskit Community. Qiskit aer v0.15.1. https://pypi.org/project/qiskit-aer/0.15.1/ (2024). Accessed: 2025-11-18. 88.The Qiskit Nature Developers and Contr...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1063/5 2020
-
[75]
Qiskit Development Team.Qiskit SDK Docs: Qiskit Circuit Library - v1.3.3. IBM Quantum (2025). Accessed: 2025-11-07
work page 2025
-
[76]
Hammes-Schiffer, S. & Tully, J. C. Proton transfer in solution: Molecular dynamics with quantum transitions.The J. Chem. Phys.101, 4657–4667, DOI: 10.1063/1.467455 (1994)
-
[77]
Tuckerman, M. E., Marx, D., Klein, M. L. & Parrinello, M. On the quantum nature of the shared proton in hydrogen bonds.Science275, 817–820, DOI: 10.1126/science.275.5301.817 (1997). 18/19
-
[78]
Morrone, J. A. & Car, R. Nuclear quantum effects in water.Phys. Rev. Lett.101, 017801, DOI: 10.1103/PhysRevLett.101. 017801 (2008)
-
[79]
Reece, S. Y . & Nocera, D. G. Proton-coupled electron transfer in biology: Results from synergistic studies in natural and model systems.Annu. Rev. Biochem.78, 673–699, DOI: https://doi.org/10.1146/annurev.biochem.78.080207.092132 (2009)
-
[80]
Li, X.-Z., Walker, B. & Michaelides, A. Quantum nature of the hydrogen bond.Proc. Natl. Acad. Sci.108, 6369–6373, DOI: 10.1073/pnas.1016653108 (2011)
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.