Ligandformer: A Graph Neural Network for Predicting Compound Property with Robust Interpretation
Pith reviewed 2026-05-24 12:20 UTC · model grok-4.3
The pith
Ligandformer integrates attention maps across graph neural network layers to link compound property predictions directly to molecular substructures.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
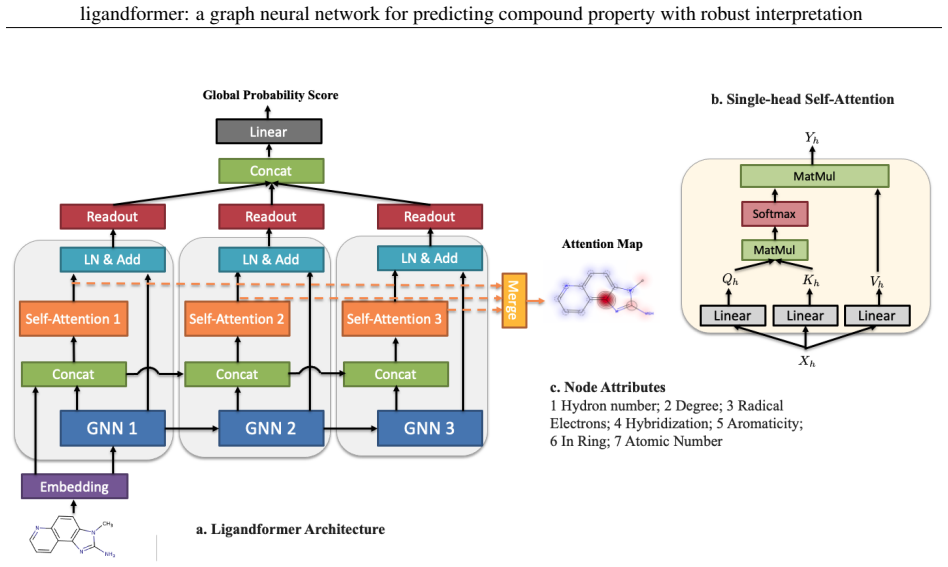
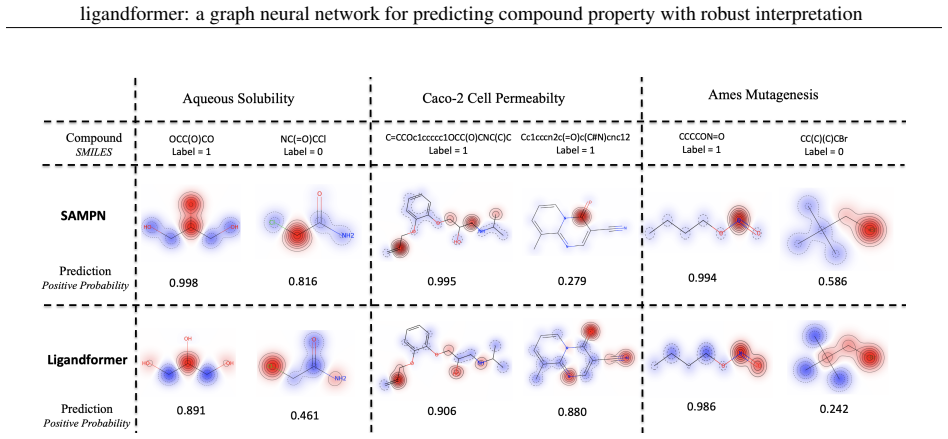
Ligandformer is a multi-layer self-attention based graph neural network framework for compound property prediction that integrates attention maps from different network blocks; the resulting map reflects the model's local interest on compound structure and indicates the relationship between the predicted property and its molecular features, while delivering robust predictions across experimental rounds and generalization to varied chemical or biological properties.
What carries the argument
The integrated attention map formed by combining self-attention outputs from multiple network blocks, which serves as a visible indicator of the relationship between predicted compound property and molecular structure.
If this is right
- Users receive both a property score and a visible structural map that can be compared against expert chemical knowledge for the same compound.
- Predictions remain consistent when the same model is retrained or evaluated in separate experimental rounds.
- The same architecture applies to multiple distinct chemical or biological properties without major redesign.
- The dual output supports direct use in structure optimization workflows by highlighting influential molecular features.
- Performance exceeds standard graph neural network baselines on accuracy, stability, and cross-property generalization.
Where Pith is reading between the lines
- If the attention maps align with established chemical rules on held-out compounds, the method could accelerate hypothesis generation for new molecule design.
- Built-in maps might reduce reliance on post-hoc interpretability techniques when applying graph models to molecular data.
- The framework could be tested on larger or more complex molecular systems to examine whether the same integration of attention maps scales to multi-component biological processes.
Load-bearing premise
The integrated attention map from different network blocks accurately and meaningfully indicates the relationship between the predicted compound property and molecular structure without requiring separate validation against chemical knowledge.
What would settle it
A test set where the model's attention maps repeatedly highlight substructures known by chemists to be irrelevant to the target property, yet the numerical predictions remain accurate.
Figures

read the original abstract
Robust and efficient interpretation of QSAR methods is quite useful to validate AI prediction rationales with subjective opinion (chemist or biologist expertise), understand sophisticated chemical or biological process mechanisms, and provide heuristic ideas for structure optimization in pharmaceutical industry. For this purpose, we construct a multi-layer self-attention based Graph Neural Network framework, namely Ligandformer, for predicting compound property with interpretation. Ligandformer integrates attention maps on compound structure from different network blocks. The integrated attention map reflects the machine's local interest on compound structure, and indicates the relationship between predicted compound property and its structure. This work mainly contributes to three aspects: 1. Ligandformer directly opens the black-box of deep learning methods, providing local prediction rationales on chemical structures. 2. Ligandformer gives robust prediction in different experimental rounds, overcoming the ubiquitous prediction instability of deep learning methods. 3. Ligandformer can be generalized to predict different chemical or biological properties with high performance. Furthermore, Ligandformer can simultaneously output specific property score and visible attention map on structure, which can support researchers to investigate chemical or biological property and optimize structure efficiently. Our framework outperforms over counterparts in terms of accuracy, robustness and generalization, and can be applied in complex system study.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes Ligandformer, a multi-layer self-attention Graph Neural Network for compound property prediction. It integrates attention maps across network blocks to generate local interpretations that purportedly reflect the model's interest in molecular substructures and thereby indicate structure-property relationships. The authors assert three contributions: direct opening of the deep-learning black box via these rationales, robust predictions across experimental rounds, and strong generalization to varied chemical/biological properties, with outperformance versus counterparts on accuracy, robustness, and generalization; the model simultaneously outputs a property score and a visible attention map.
Significance. If the performance claims are substantiated with proper benchmarks and the attention maps are shown to align with chemical causality, the work could meaningfully advance interpretable QSAR modeling for drug discovery by combining prediction with built-in structural rationales.
major comments (2)
- [Abstract] Abstract: the assertion that Ligandformer 'outperforms over counterparts in terms of accuracy, robustness and generalization' is presented without any reported metrics, datasets, experimental protocols, baselines, or error analysis, leaving the three enumerated contributions without visible empirical support.
- [Abstract] Abstract: the central claim that the integrated attention map 'indicates the relationship between predicted compound property and its structure' rests on the untested assumption that multi-block self-attention faithfully highlights causally relevant atoms or substructures; no ablation against gradient/perturbation attributions, no comparison to known SAR motifs on benchmark molecules, and no expert-agreement metric are described.
Simulated Author's Rebuttal
We thank the referee for the constructive comments. We address each point below and propose targeted revisions to the abstract to better ground the claims with available evidence from the full manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the assertion that Ligandformer 'outperforms over counterparts in terms of accuracy, robustness and generalization' is presented without any reported metrics, datasets, experimental protocols, baselines, or error analysis, leaving the three enumerated contributions without visible empirical support.
Authors: The abstract serves as a concise overview; the full manuscript reports detailed benchmarks across multiple datasets (including performance tables, baseline comparisons such as standard GNNs, and analyses of robustness over experimental rounds plus generalization to varied properties). To improve self-containment, we will revise the abstract to incorporate representative quantitative results and reference the experimental protocols. revision: yes
-
Referee: [Abstract] Abstract: the central claim that the integrated attention map 'indicates the relationship between predicted compound property and its structure' rests on the untested assumption that multi-block self-attention faithfully highlights causally relevant atoms or substructures; no ablation against gradient/perturbation attributions, no comparison to known SAR motifs on benchmark molecules, and no expert-agreement metric are described.
Authors: The attention integration is presented as capturing the model's learned local focus on substructures via the self-attention layers. We agree the original wording overstates the causal link; the manuscript does not contain the suggested ablations or expert metrics. We will revise the abstract to state that the maps reflect the model's structural interest for interpretation purposes, without asserting direct indication of causal relationships. revision: partial
Circularity Check
No circularity in Ligandformer derivation or claims
full rationale
The paper proposes a multi-layer self-attention GNN architecture (Ligandformer) whose core outputs—property predictions and integrated attention maps—are direct consequences of the model design and training process rather than any self-referential reduction. No equations, derivations, or parameter-fitting steps are described that equate the claimed interpretation, robustness, or generalization to inputs by construction. Claims of 'opening the black-box' rest on the architectural feature of attention integration, not on a loop where the output is presupposed in the definition. No load-bearing self-citations or uniqueness theorems appear in the provided text. The framework is self-contained as an empirical modeling contribution.
Axiom & Free-Parameter Ledger
Forward citations
Cited by 2 Pith papers
-
Do Larger Models Really Win in Drug Discovery? A Benchmark Assessment of Model Scaling in AI-Driven Molecular Property and Activity Prediction
A benchmark across 156 comparisons finds classical ML models win 116 times while larger pretrained and LLM models win far fewer, showing predictive performance depends on model-task fit rather than scale.
-
Do Larger Models Really Win in Drug Discovery? A Benchmark Assessment of Model Scaling in AI-Driven Molecular Property and Activity Prediction
Large benchmark shows classical ML and GNNs outperform pretrained large models on most of 22 drug-discovery endpoints under strict cross-validation.
Reference graph
Works this paper leans on
-
[1]
Moleculenet: a benchmark for molecular machine learning
Zhenqin Wu, Bharath Ramsundar, Evan N Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S Pappu, Karl Leswing, and Vijay Pande. Moleculenet: a benchmark for molecular machine learning. Chemical science, 9(2): 513–530, 2018
work page 2018
-
[2]
Step change improvement in admet prediction with potentialnet deep featurization
EN Feinberg, R Sheridan, E Joshi, VS Pande, and AC Cheng. Step change improvement in admet prediction with potentialnet deep featurization. arxiv. org, 2019
work page 2019
-
[3]
Qsar studies of fabh inhibitors using graph theoretical & quantum chemical descriptors
Dipanjan Sarkar, Shyamal Sharma, Subhasis Mukhopadhyay, and Asim Kumar Bothra. Qsar studies of fabh inhibitors using graph theoretical & quantum chemical descriptors. Pharmacophore, 7(4), 2016
work page 2016
-
[4]
Zheng Shao, Yuya Hirayama, Yoshihiro Yamanishi, and Hiroto Saigo. Mining discriminative patterns from graph data with multiple labels and its application to quantitative structure–activity relationship (qsar) models. Journal of chemical information and modeling, 55(12):2519–2527, 2015
work page 2015
-
[5]
Molecule property prediction based on spatial graph embedding
Xiaofeng Wang, Zhen Li, Mingjian Jiang, Shuang Wang, Shugang Zhang, and Zhiqiang Wei. Molecule property prediction based on spatial graph embedding. Journal of chemical information and modeling, 59(9):3817–3828, 2019
work page 2019
-
[6]
Chemi-net: a molecular graph convolutional network for accurate drug property prediction
Ke Liu, Xiangyan Sun, Lei Jia, Jun Ma, Haoming Xing, Junqiu Wu, Hua Gao, Yax Sun, Florian Boulnois, and Jie Fan. Chemi-net: a molecular graph convolutional network for accurate drug property prediction. International journal of molecular sciences, 20(14):3389, 2019
work page 2019
-
[7]
Predicting activities without computing descriptors: graph machines for qsar
A Goulon, T Picot, A Duprat, and G Dreyfus. Predicting activities without computing descriptors: graph machines for qsar. SAR and QSAR in Environmental Research, 18(1-2):141–153, 2007
work page 2007
-
[8]
Bowen Tang, Skyler T Kramer, Meijuan Fang, Yingkun Qiu, Zhen Wu, and Dong Xu. A self-attention based message passing neural network for predicting molecular lipophilicity and aqueous solubility. Journal of cheminformatics, 12(1):1–9, 2020
work page 2020
-
[9]
Artem Cherkasov, Eugene N Muratov, Denis Fourches, Alexandre Varnek, Igor I Baskin, Mark Cronin, John Dearden, Paola Gramatica, Yvonne C Martin, Roberto Todeschini, et al. Qsar modeling: where have you been? where are you going to? Journal of medicinal chemistry, 57(12):4977–5010, 2014
work page 2014
-
[10]
The rise of deep learning in drug discovery
Hongming Chen, Ola Engkvist, Yinhai Wang, Marcus Olivecrona, and Thomas Blaschke. The rise of deep learning in drug discovery. Drug discovery today, 23(6):1241–1250, 2018
work page 2018
-
[11]
Benchmarks for interpretation of qsar models.Journal of cheminformatics, 13(1):1–20, 2021
Mariia Matveieva and Pavel Polishchuk. Benchmarks for interpretation of qsar models.Journal of cheminformatics, 13(1):1–20, 2021
work page 2021
-
[12]
Inductive Representation Learning on Large Graphs
William L Hamilton, Rex Ying, and Jure Leskovec. Inductive representation learning on large graphs. arXiv preprint arXiv:1706.02216, 2017
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[13]
How Powerful are Graph Neural Networks?
Keyulu Xu, Weihua Hu, Jure Leskovec, and Stefanie Jegelka. How powerful are graph neural networks? arXiv preprint arXiv:1810.00826, 2018
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[14]
Weisfeiler and leman go neural: Higher-order graph neural networks
Christopher Morris, Martin Ritzert, Matthias Fey, William L Hamilton, Jan Eric Lenssen, Gaurav Rattan, and Martin Grohe. Weisfeiler and leman go neural: Higher-order graph neural networks. In Proceedings of the AAAI Conference on Artificial Intelligence, volume 33, pages 4602–4609, 2019
work page 2019
-
[15]
Asap: Adaptive structure aware pooling for learning hierarchical graph representations
Ekagra Ranjan, Soumya Sanyal, and Partha Talukdar. Asap: Adaptive structure aware pooling for learning hierarchical graph representations. In Proceedings of the AAAI Conference on Artificial Intelligence, volume 34, pages 5470–5477, 2020
work page 2020
-
[16]
Mahesh Pal. Random forest classifier for remote sensing classification.International journal of remote sensing, 26(1):217–222, 2005
work page 2005
-
[17]
What is a support vector machine? Nature biotechnology, 24(12):1565–1567, 2006
William S Noble. What is a support vector machine? Nature biotechnology, 24(12):1565–1567, 2006
work page 2006
-
[18]
Smiles, a chemical language and information system
David Weininger. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. Journal of chemical information and computer sciences, 28(1):31–36, 1988
work page 1988
-
[19]
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Łukasz Kaiser, and Illia Polosukhin. Attention is all you need. Advances in neural information processing systems, 30, 2017
work page 2017
-
[20]
Language models are few-shot learners
Tom Brown, Benjamin Mann, Nick Ryder, Melanie Subbiah, Jared D Kaplan, Prafulla Dhariwal, Arvind Nee- lakantan, Pranav Shyam, Girish Sastry, Amanda Askell, et al. Language models are few-shot learners. Advances in neural information processing systems, 33:1877–1901, 2020
work page 1901
-
[21]
Exploring the Limits of Transfer Learning with a Unified Text-to-Text Transformer
Colin Raffel, Noam Shazeer, Adam Roberts, Katherine Lee, Sharan Narang, Michael Matena, Yanqi Zhou, Wei Li, and Peter J Liu. Exploring the limits of transfer learning with a unified text-to-text transformer. arXiv preprint arXiv:1910.10683, 2019. 6 ligandformer: a graph neural network for predicting compound property with robust interpretation
work page internal anchor Pith review Pith/arXiv arXiv 1910
-
[22]
Niki Parmar, Ashish Vaswani, Jakob Uszkoreit, Lukasz Kaiser, Noam Shazeer, Alexander Ku, and Dustin Tran. Image transformer. In International Conference on Machine Learning, pages 4055–4064. PMLR, 2018
work page 2018
-
[23]
End-to-end object detection with transformers
Nicolas Carion, Francisco Massa, Gabriel Synnaeve, Nicolas Usunier, Alexander Kirillov, and Sergey Zagoruyko. End-to-end object detection with transformers. In European conference on computer vision , pages 213–229. Springer, 2020
work page 2020
-
[24]
Efficient transformers: A survey
Yi Tay, Mostafa Dehghani, Dara Bahri, and Donald Metzler. Efficient transformers: A survey. arXiv preprint arXiv:2009.06732, 2020
-
[25]
Rdkit: A software suite for cheminformatics, computational chemistry, and predictive modeling, 2013
Greg Landrum et al. Rdkit: A software suite for cheminformatics, computational chemistry, and predictive modeling, 2013
work page 2013
-
[26]
Bharath Ramsundar, Peter Eastman, Patrick Walters, and Vijay Pande. Deep learning for the life sciences: applying deep learning to genomics, microscopy, drug discovery, and more. O’Reilly Media, 2019
work page 2019
-
[27]
Analyzing learned molecular representations for property prediction
Kevin Yang, Kyle Swanson, Wengong Jin, Connor Coley, Philipp Eiden, Hua Gao, Angel Guzman-Perez, Timothy Hopper, Brian Kelley, Miriam Mathea, et al. Analyzing learned molecular representations for property prediction. Journal of chemical information and modeling, 59(8):3370–3388, 2019
work page 2019
-
[28]
Jimmy Lei Ba, Jamie Ryan Kiros, and Geoffrey E Hinton. Layer normalization. arXiv preprint arXiv:1607.06450, 2016
work page internal anchor Pith review Pith/arXiv arXiv 2016
-
[29]
Petar Veliˇckovi´c, Guillem Cucurull, Arantxa Casanova, Adriana Romero, Pietro Lio, and Yoshua Bengio. Graph attention networks. arXiv preprint arXiv:1710.10903, 2017
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[30]
Semi-Supervised Classification with Graph Convolutional Networks
Thomas N Kipf and Max Welling. Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907, 2016
work page internal anchor Pith review Pith/arXiv arXiv 2016
-
[31]
Matt W Gardner and SR Dorling. Artificial neural networks (the multilayer perceptron)—a review of applications in the atmospheric sciences. Atmospheric environment, 32(14-15):2627–2636, 1998
work page 1998
-
[32]
Krzysztof C Kiwiel. Convergence and efficiency of subgradient methods for quasiconvex minimization.Mathe- matical programming, 90(1):1–25, 2001
work page 2001
-
[33]
Adam: A Method for Stochastic Optimization
Diederik P Kingma and Jimmy Ba. Adam: A method for stochastic optimization. arXiv preprint arXiv:1412.6980, 2014
work page internal anchor Pith review Pith/arXiv arXiv 2014
-
[34]
Adabelief optimizer: Adapting stepsizes by the belief in observed gradients
Juntang Zhuang, Tommy Tang, Sekhar Tatikonda, Nicha Dvornek, Yifan Ding, Xenophon Papademetris, and James S Duncan. Adabelief optimizer: Adapting stepsizes by the belief in observed gradients. arXiv preprint arXiv:2010.07468, 2020
-
[35]
Iurii Sushko, Sergii Novotarskyi, Robert Körner, Anil Kumar Pandey, Matthias Rupp, Wolfram Teetz, Stefan Brandmaier, Ahmed Abdelaziz, V olodymyr V Prokopenko, Vsevolod Y Tanchuk, et al. Online chemical modeling environment (ochem): web platform for data storage, model development and publishing of chemical information. Journal of computer-aided molecular ...
work page 2011
-
[36]
Caco-2 cell permeability assays to measure drug absorption
Richard B van Breemen and Yongmei Li. Caco-2 cell permeability assays to measure drug absorption. Expert opinion on drug metabolism & toxicology, 1(2):175–185, 2005
work page 2005
-
[37]
Chemical information for chemists: a primer
Judith Currano and Dana Roth. Chemical information for chemists: a primer. Royal Society of Chemistry, 2014
work page 2014
-
[38]
The ames salmonella/microsome mutagenicity assay
Kristien Mortelmans and Errol Zeiger. The ames salmonella/microsome mutagenicity assay. Mutation re- search/fundamental and molecular mechanisms of mutagenesis, 455(1-2):29–60, 2000. 7
work page 2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.