Atom-anchored LLMs speak Chemistry: A Retrosynthesis Demonstration
Pith reviewed 2026-05-22 11:56 UTC · model grok-4.3
The pith
General-purpose LLMs perform single-step retrosynthesis by anchoring reasoning to unique atomic identifiers without task-specific training
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
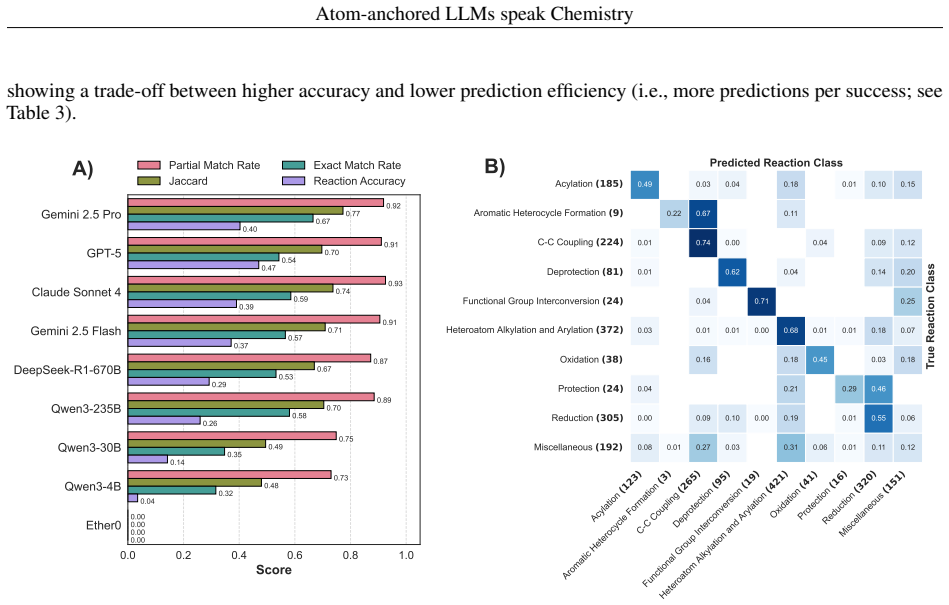
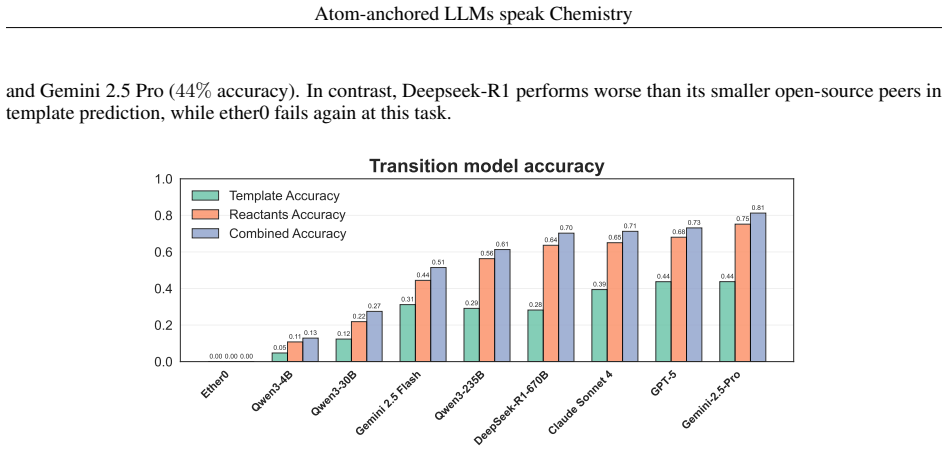
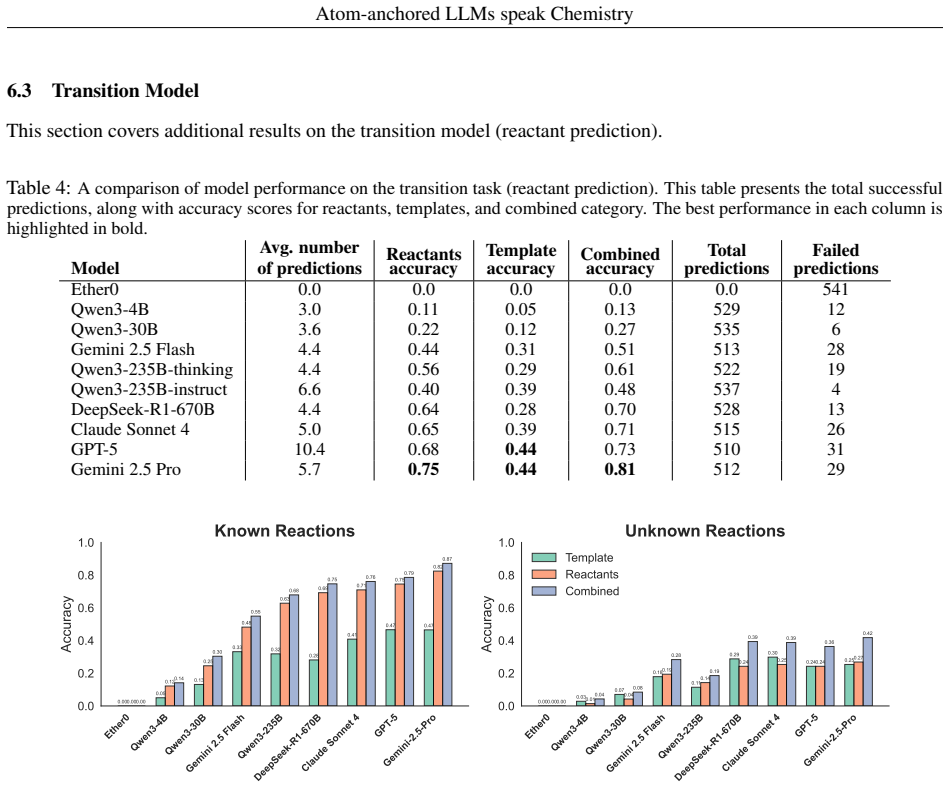
By assigning unique atomic identifiers to molecules, general-purpose LLMs can perform chain-of-thought reasoning that first locates chemically relevant fragments and their associated transformation classes and then, when supplied with class examples, predicts the reactant molecules that would produce the target in a single retrosynthetic step, achieving success rates of at least 90 percent for reaction-site identification, 40 percent for named reaction classes, and 74 percent for final reactant prediction across benchmarks and real drug-discovery compounds without any task-specific training.
What carries the argument
Atom-anchored chain-of-thought reasoning, in which unique atomic identifiers are attached to the molecular graph so the LLM can reference specific atoms when identifying fragments and transformation classes.
If this is right
- LLMs can reach competitive performance on retrosynthesis benchmarks using only prompting rather than fine-tuning.
- The same anchoring technique supplies a reusable blueprint for other molecular-reasoning tasks that involve identifying sites and transformations.
- Drug-discovery workflows can incorporate general-purpose LLMs to propose reactant sets for newly designed target molecules.
- Performance on named reaction classes improves when the second step supplies concrete class examples.
Where Pith is reading between the lines
- The method could be chained across multiple steps to tackle full retrosynthetic routes by reusing the same atomic identifiers at each stage.
- Because no domain-specific weights are updated, the approach lowers the barrier for labs that lack large GPU clusters to experiment with molecular reasoning.
- Similar identifier-anchoring might be tested on other structured scientific objects such as protein binding sites or crystal lattices.
Load-bearing premise
That supplying unique atomic identifiers is enough for a general-purpose LLM to perform chemically valid molecular reasoning and transformation prediction without any chemistry-specific training or fine-tuning.
What would settle it
Running the same prompts on a fresh set of expert-validated molecules but with the atomic identifiers removed or randomized and observing whether reactant-prediction accuracy drops below 50 percent.
Figures











read the original abstract
Applications of machine learning in chemistry are often limited by the scarcity and expense of labeled data, restricting traditional supervised methods. In this work, we introduce a framework for molecular reasoning using general-purpose Large Language Models (LLMs) that operates without requiring task-specific model training. Our method anchors chain-of-thought reasoning to the molecular structure by using unique atomic identifiers. First, the LLM performs a zero-shot task to identify relevant fragments and their associated chemical labels or transformation classes. In an optional second step, this position-aware information is used in a few-shot task with provided class examples to predict the chemical transformation. We apply our framework to single-step retrosynthesis, a task where LLMs have previously underperformed. Across academic benchmarks and expert-validated drug discovery molecules, our work enables LLMs to achieve high success rates in identifying chemically plausible reaction sites ($\geq90\%$), named reaction classes ($\geq40\%$), and final reactants ($\geq74\%$). Ultimately, our work establishes a general blueprint for applying LLMs to challenges where molecular reasoning and molecular transformations are key, positioning atom-anchored LLMs as a powerful solution for data-scarce chemistry domains.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents a framework for single-step retrosynthesis using general-purpose LLMs without task-specific fine-tuning. It anchors chain-of-thought reasoning to molecular structures via unique atomic identifiers, first identifying fragments and reaction classes in zero-shot mode, then optionally using few-shot examples to predict transformations. The central empirical claim is that this enables LLMs to achieve ≥90% success on chemically plausible reaction sites, ≥40% on named reaction classes, and ≥74% on final reactants across academic benchmarks and expert-validated drug-discovery molecules.
Significance. If the reported success rates are reproducible and the atomic-anchoring mechanism is shown to be causal, the work would supply a practical, training-free blueprint for LLM-based molecular reasoning in data-scarce chemistry domains. The absence of any free parameters or invented entities in the reported approach is a methodological strength that distinguishes it from supervised retrosynthesis models.
major comments (2)
- [Methods / Results] The manuscript provides no ablation that isolates the effect of the unique atomic identifiers. A direct comparison of anchored versus standard SMILES prompts on identical molecules and tasks is required to establish that the identifiers, rather than pre-trained reaction knowledge or prompt phrasing, drive the reported accuracies (≥90% sites, ≥40% classes, ≥74% reactants). Without this control the central claim that atom-anchoring enables chemically valid zero-shot/few-shot retrosynthesis remains unproven.
- [Abstract / Results] The abstract and results sections state success rates but supply no information on experimental protocol, baseline comparisons, error bars, dataset sizes, or how chemically plausible reactants were validated by experts. These omissions prevent assessment of whether the empirical claims are statistically supported or reproducible.
minor comments (2)
- [Methods] Clarify the exact format of the atomic identifiers and how they are injected into the LLM prompt (e.g., SMILES augmentation syntax).
- [Methods] Specify the number of few-shot examples used in the optional second step and whether performance varies with example count.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. The comments help clarify the need for stronger evidence on the role of atomic identifiers and for greater transparency in reporting experimental details. We respond to each major comment below and will revise the manuscript to address them where possible.
read point-by-point responses
-
Referee: [Methods / Results] The manuscript provides no ablation that isolates the effect of the unique atomic identifiers. A direct comparison of anchored versus standard SMILES prompts on identical molecules and tasks is required to establish that the identifiers, rather than pre-trained reaction knowledge or prompt phrasing, drive the reported accuracies (≥90% sites, ≥40% classes, ≥74% reactants). Without this control the central claim that atom-anchoring enables chemically valid zero-shot/few-shot retrosynthesis remains unproven.
Authors: We agree that an explicit ablation comparing anchored prompts against standard SMILES prompts on the same molecules and tasks would strengthen the causal argument. The current manuscript demonstrates the overall framework performance but does not include this head-to-head control. In the revised version we will add the requested ablation across the benchmark sets and expert-validated molecules, reporting success rates for both zero-shot fragment identification and few-shot reactant prediction. This addition will directly test whether the identifiers, rather than general pre-trained knowledge, account for the observed accuracies. revision: yes
-
Referee: [Abstract / Results] The abstract and results sections state success rates but supply no information on experimental protocol, baseline comparisons, error bars, dataset sizes, or how chemically plausible reactants were validated by experts. These omissions prevent assessment of whether the empirical claims are statistically supported or reproducible.
Authors: The full manuscript contains a Methods section that specifies the datasets (academic benchmarks and expert-curated drug-like molecules), the number of molecules evaluated, the expert validation protocol for chemical plausibility, and the prompting procedure. Error bars and statistical details appear in the Results figures and tables. To address the referee’s concern about accessibility, we will revise the abstract to include a concise statement of dataset scale and validation approach, and we will add explicit cross-references in the Results section to the Methods details on reproducibility. We will also ensure baseline comparisons (where performed) and error reporting are highlighted more clearly. revision: partial
Circularity Check
No significant circularity: empirical demonstration on external benchmarks
full rationale
The paper introduces an atom-anchored prompting framework for zero-shot and few-shot retrosynthesis with general-purpose LLMs and reports empirical success rates (≥90% reaction sites, ≥40% named classes, ≥74% reactants) on academic benchmarks plus expert-validated drug discovery molecules. These metrics are measured against independent external test sets rather than quantities defined by the authors' own fitted parameters or self-referential equations. No derivation chain, mathematical ansatz, or uniqueness theorem is presented that reduces the central claims to inputs by construction. The work is self-contained as an empirical demonstration; any self-citations that may exist are not load-bearing for the reported accuracies.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption General-purpose LLMs can perform chemically valid molecular reasoning when chain-of-thought is anchored to unique atomic identifiers
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Our method anchors chain-of-thought reasoning to the molecular structure by using unique atomic identifiers... zero-shot task to identify relevant fragments and their associated chemical labels or transformation classes... few-shot task with provided class examples to predict the chemical transformation.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
DeepSeek-R1: Incentivizing Reasoning Capability in LLMs via Reinforcement Learning
ISSN 1758-2946. doi:10.1186/s13321-025-00986-6. Seongmin Kim, Yousung Jung, and Joshua Schrier. Large Language Models for Inorganic Synthesis Predictions, June 2024. Joseph M. Cavanagh, Kunyang Sun, Andrew Gritsevskiy, Dorian Bagni, Thomas D. Bannister, and Teresa Head-Gordon. SmileyLlama: Modifying Large Language Models for Directed Chemical Space Explor...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1186/s13321-025-00986-6 2024
-
[2]
ISSN 0022-2623, 1520-4804. doi:10.1021/acs.jmedchem.6b01482. Xiaoting Li, Hao Chang, Jara Bouma, Laura V . De Paus, Partha Mukhopadhyay, Janos Paloczi, Mohammed Mustafa, Cas Van Der Horst, Sanjay Sunil Kumar, Lijie Wu, Yanan Yu, Richard J. B. H. N. Van Den Berg, Antonius P. A. Janssen, Aron Lichtman, Zhi-Jie Liu, Pal Pacher, Mario Van Der Stelt, Laura H. ...
-
[3]
**Step 1: Identify All Candidate Transformations** 15Process steps A - L sequentially. For each step, you must perform a complete and independent analysis to identify all transformations that fit its description. A finding in one step does not exclude findings in others. 16* **Input:** The ‘product_smiles ‘. 17* **Process:** 18* A) **Symmetry Analysis:** ...
-
[4]
**Step 2: Assign Candidate Reactions** 35* **Input:** The list of transformation strings from Step 1. 36* **Process:** For each transformation, determine all appropriate forward reaction names. A single transformation may have multiple corresponding reactions. 37* **Output (Internal):** A list of objects, where each object contains a transformation and a ...
-
[5]
**Step 3: Expand and Evaluate Pairs** 41* **Input:** The list of objects from Step 2. 42* **Process:** Expand the input into a flat list by creating a **new, separate entry for each reaction** associated with a transformation. Then, for each of these new entries, apply the Retrosynthetic Analysis Framework to assign a ‘Retrosynthesis Importance‘ value and...
-
[6]
**Step 4: Final Formatting and Priority Assignment** 46* **Input:** The flat list of objects from Step 3. 47* **Process:** For each object, format it according to the ‘Constraints & Formatting Rules‘. Then, calculate a ‘ Priority‘ number for each entry by ranking them based on two criteria: 1. ‘"isInOntology"‘ (‘true‘ before ‘false ‘), and 2. ‘"Retrosynth...
-
[7]
23* **Process:** If a ‘forward_reaction_name‘ is provided, use it as the sole reaction
**Step 1: Determine Reaction(s) to Model** 22* **Input:** The ‘forward_reaction_name‘ (optional) and ‘reaction_center_atoms‘ from the user. 23* **Process:** If a ‘forward_reaction_name‘ is provided, use it as the sole reaction. If not, analyze the ‘ reaction_center_atoms‘ to generate a list of potential ‘forward_reaction_name‘s. 24* **Output (Internal):**...
-
[8]
**Step 2: Refine Reaction Center** 27* **Input:** The list of ‘forward_reaction_name‘s (Step 1), the users ‘reaction_center_atoms‘, and any ‘ retrosynthesis_reaction_examples ‘. 28* **Process:** For each ‘forward_reaction_name‘, use your expert chemical knowledge and the provided examples to determine the **precise and complete reaction center**. The user...
-
[9]
**Step 3: Extract Atom-Level Reaction Template** 32* **Input:** The list of ‘forward_reaction_name‘s from Step 1, the **precise reaction center** from step 2, and the user-provided ‘retrosynthesis_reaction_examples ‘. 33* **Process:** For each ‘forward_reaction_name‘, analyze its corresponding valid example(s). Your primary goal is to extract the **struct...
-
[10]
**Step 4: Generate Precursor Molecule(s)** 95* **Input:** The ‘product_smiles‘ and ‘precise_reaction_center_atoms ‘. 96* **Process:** Based on the number of fragments implied by the transformation type (e.g., two for an intermolecular disconnection, one for an FGI, three for a 3-component MCR), generate the corresponding core precursor molecule(s ). This ...
-
[11]
**Step 5: Apply Reaction Template to Generate Reactant Permutations** 100* **Input:** The precursor(s) (Step 4) and the reaction templates (Step 3). 101* **Process:** For each reactions template, apply the extracted retrosynthetic template to the precursor(s). The ‘ precise_reaction_center_atoms‘ provided by the user defines the **locality** of the transf...
-
[12]
**Fragment-Role Permutations:** For a disconnection into multiple fragments with distinct reactive groups, you must generate reactant sets for **all** possible assignments of those groups to the fragments
-
[13]
**Intra-Group Class Permutations:** If a generated reactive group belongs to a general chemical class (e.g., an "organohalide," "leaving␣group," or "protecting␣group"), you are required to generate an exhaustive list of separate options for **all chemically distinct members of that class known to be compatible with the reaction.** 104The model is **explic...
-
[14]
109* **Process:** For each generated option, perform a rigorous chemical validation
**Step 6: Validate and Justify Each Option** 108* **Input:** The list of potential reactant options from Step 5. 109* **Process:** For each generated option, perform a rigorous chemical validation. 110* A) **Stability:** Are the proposed reactants chemically stable? 111* B) **Chemoselectivity:** Would the reaction be selective? Are there other functional ...
-
[15]
**Group Options:** Begin by grouping the list of validated options by their ‘forward_reaction_name ‘
-
[16]
It signals that a member of this chemical class (e.g
**Extract Wildcard Reaction Class** Looking at the validated options and their reaction names, you must deduct a general reaction class template if possible using the ‘<CLASS:..>‘ tag. It signals that a member of this chemical class (e.g. ‘<CLASS:AmineProtectingGroup >‘) should be used instead of an explicit molecular structure
-
[17]
**Generate General Template Entry (if applicable):** For each extracted general reaction class template, you ** should** create one additional, special permutation object derived from the two provided general reaction classes . This object serves as the general, machine-readable representation for the entire transformation class and should be placed at th...
-
[18]
**Assemble Final List:** For each unique reaction, create a single object containing the ‘forward_reaction_name‘ and its final ‘reactant_permutations‘ list. This list will now contain the general template entry at the top (if applicable), followed by all the validated, specific examples from Step 6
-
[19]
**Finalize and Clean:** Assemble these grouped objects into the final ‘reaction_analysis‘ list according to the ‘ Output Schema‘. Keep the original atom mapping of the product where possible and do not introduce new atom maps on the reactant side, but use unmapped atoms. 131* **Output:** The final, single JSON object. 132 133**Output Schema - Strict JSON ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.