MolClaw: An Autonomous Agent with Hierarchical Skills for Drug Molecule Evaluation, Screening, and Optimization
Pith reviewed 2026-05-21 10:42 UTC · model grok-4.3
The pith
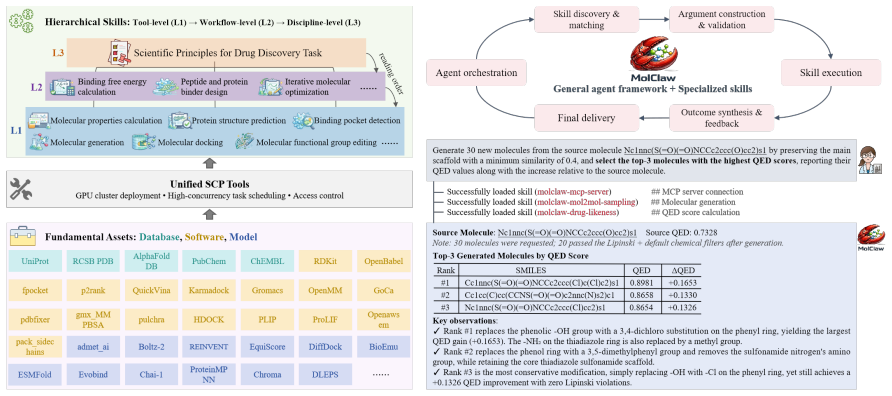
MolClaw uses a three-tier skill hierarchy to orchestrate dozens of tools for drug molecule evaluation, screening, and optimization.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
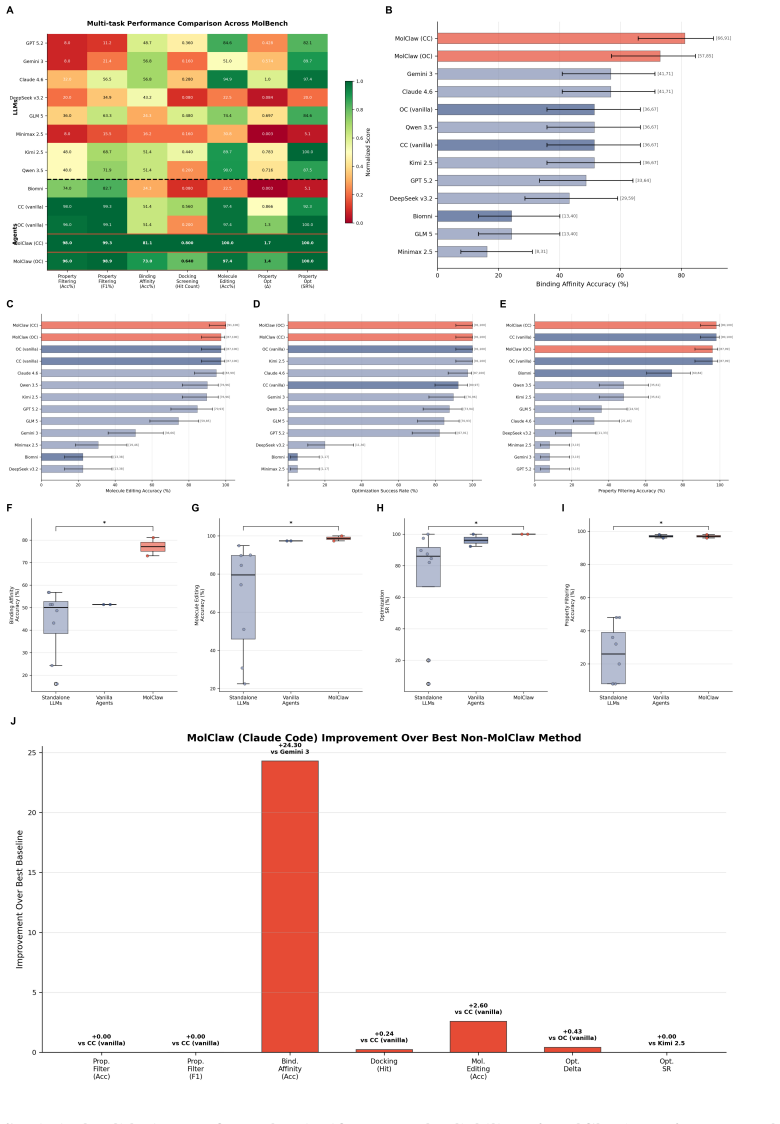
MolClaw unifies over 30 specialized domain resources through a three-tier hierarchical skill architecture comprising 70 skills in total. Tool-level skills standardize atomic operations, workflow-level skills compose them into validated pipelines that include quality checks and reflection, and a discipline-level skill supplies scientific principles that govern planning and verification across scenarios. The paper introduces MolBench, a benchmark spanning molecular screening, optimization, and end-to-end discovery challenges that require 8 to 50 or more sequential tool calls. On this benchmark MolClaw records state-of-the-art performance across all metrics. Ablation studies indicate that the性能
What carries the argument
Three-tier hierarchical skill architecture that separates atomic tool operations, composable workflow pipelines with reflection, and discipline-level scientific principles for consistent planning and verification.
If this is right
- Workflow orchestration competence, rather than access to individual tools, becomes the primary bottleneck for AI-driven drug discovery.
- Agents that incorporate workflow-level skills with quality checks maintain higher success rates across long sequences of 20 to 50 tool calls.
- Discipline-level skills improve consistency of planning and verification without requiring task-specific retraining.
- Performance differences between hierarchical and flat agents concentrate on complex structured problems and vanish on tasks solvable by direct scripting.
Where Pith is reading between the lines
- The same tiered skill structure could be tested in adjacent domains such as materials screening or synthetic biology that also rely on chained experimental tools.
- Benchmarks that add longer sequences or integration with actual laboratory execution would provide stronger tests of whether the hierarchy scales beyond simulation.
- The discipline-level layer suggests a route to transfer scientific constraints across related molecular tasks without rebuilding lower-level skills.
Load-bearing premise
The MolBench tasks and evaluation metrics are representative of real drug-discovery difficulty, and the reported performance advantage is caused by the three-tier hierarchical skill design rather than other unstated implementation choices.
What would settle it
An ablation on MolBench that disables workflow-level and discipline-level skills and measures whether success rates on tasks requiring 20 or more tool calls fall to the level of simple ad-hoc scripting baselines.
Figures







read the original abstract
Computational drug discovery, particularly the complex workflows of drug molecule screening and optimization, requires orchestrating dozens of specialized tools in multi-step workflows, yet current AI agents struggle to maintain robust performance and consistently underperform in these high-complexity scenarios. Here we present MolClaw, an autonomous agent that leads drug molecule evaluation, screening, and optimization. It unifies over 30 specialized domain resources through a three-tier hierarchical skill architecture (70 skills in total) that facilitates agent long-term interaction at runtime: tool-level skills standardize atomic operations, workflow-level skills compose them into validated pipelines with quality check and reflection, and a discipline-level skill supplies scientific principles governing planning and verification across all scenarios in the field. Additionally, we introduce MolBench, a benchmark comprising molecular screening, optimization, and end-to-end discovery challenges spanning 8 to 50+ sequential tool calls. MolClaw achieves state-of-the-art performance across all metrics, and ablation studies confirm that gains concentrate on tasks that demand structured workflows while vanishing on those solvable with ad hoc scripting, establishing workflow orchestration competence as the primary capability bottleneck for AI-driven drug discovery.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents MolClaw, an autonomous agent with a three-tier hierarchical skill architecture (tool-level, workflow-level, and discipline-level skills, totaling 70) designed to orchestrate complex multi-step workflows for drug molecule evaluation, screening, and optimization. It unifies over 30 specialized domain resources and introduces MolBench, a benchmark consisting of molecular screening, optimization, and end-to-end discovery tasks that require 8 to 50+ sequential tool calls. The central claims are that MolClaw achieves state-of-the-art performance across all metrics on MolBench and that ablation studies demonstrate the performance gains concentrate on tasks demanding structured workflows, thereby establishing workflow orchestration competence as the primary bottleneck for AI-driven drug discovery.
Significance. If the performance claims and ablation results hold under rigorous controls, the work could meaningfully advance the design of hierarchical agents for long-horizon scientific workflows in chemistry. The MolBench benchmark may also provide a reusable testbed for evaluating agent robustness on tasks with high sequential complexity. The explicit framing of workflow orchestration as a distinct capability bottleneck is a useful conceptual contribution.
major comments (2)
- [Abstract] Abstract: the manuscript asserts SOTA results across all metrics and attributes them to the three-tier architecture via ablations, yet the abstract (and by extension the reported results) supplies no numerical scores, error bars, dataset sizes, or statistical tests. This absence makes it impossible to evaluate the magnitude or reliability of the claimed performance advantage.
- [Ablation studies] Ablation studies: the claim that gains concentrate on structured-workflow tasks while vanishing on ad-hoc ones is load-bearing for the central architectural argument. However, it is not shown whether the non-hierarchical control agents retain the full set of 70 skills, employ identical tool interfaces, or receive equivalent reflection/quality-check prompts. Without these controls, the measured advantage cannot be confidently attributed to the tiered organization rather than differences in total skill count or implementation details.
minor comments (2)
- [Introduction] Clarify the precise mapping between the stated 'over 30 specialized domain resources' and the final count of 70 skills.
- [MolBench] MolBench section: provide explicit definitions of the evaluation metrics and a clearer justification that the chosen tasks and sequential lengths are representative of real drug-discovery difficulty.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed feedback. We address each major comment below and have revised the manuscript to improve clarity and experimental transparency.
read point-by-point responses
-
Referee: [Abstract] Abstract: the manuscript asserts SOTA results across all metrics and attributes them to the three-tier architecture via ablations, yet the abstract (and by extension the reported results) supplies no numerical scores, error bars, dataset sizes, or statistical tests. This absence makes it impossible to evaluate the magnitude or reliability of the claimed performance advantage.
Authors: We agree that the abstract would benefit from explicit numerical results to allow readers to assess the scale of the reported gains. In the revised manuscript we have added representative performance figures (success rate and average tool calls on MolBench), noted the dataset sizes, and referenced the error bars and statistical comparisons that appear in the results section. revision: yes
-
Referee: [Ablation studies] Ablation studies: the claim that gains concentrate on structured-workflow tasks while vanishing on ad-hoc ones is load-bearing for the central architectural argument. However, it is not shown whether the non-hierarchical control agents retain the full set of 70 skills, employ identical tool interfaces, or receive equivalent reflection/quality-check prompts. Without these controls, the measured advantage cannot be confidently attributed to the tiered organization rather than differences in total skill count or implementation details.
Authors: We acknowledge that the ablation description did not explicitly document these controls. The non-hierarchical baselines were in fact given the identical set of 70 skills, the same tool interfaces, and equivalent reflection and quality-check prompts. To eliminate any ambiguity we have inserted a new paragraph in the ablation studies section that states these equivalences and lists the precise prompt templates used for the control agents. revision: yes
Circularity Check
No circularity: empirical claims rest on independent benchmark and ablations
full rationale
The paper introduces MolClaw's three-tier skill hierarchy and the separate MolBench benchmark, then reports empirical SOTA results plus ablation outcomes showing differential gains on structured vs. ad-hoc tasks. No equations, fitted parameters, or self-referential definitions appear in the provided text that would reduce the performance claims to the architecture by construction. The derivation chain is a standard empirical pipeline (new method + new test set + controlled comparisons) rather than a closed loop where outputs are forced by input definitions or self-citations. The ablation critique concerns experimental controls, not circular reduction of results to inputs.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Mark James Abraham, Teemu Murtola, Roland Schulz, Szilárd Páll, Jeremy C Smith, Berk Hess, and Erik Lindahl. Gromacs: High performance molecular simulations through multi-level paral- lelism from laptops to supercomputers.SoftwareX, 1:19–25, 2015
work page 2015
-
[2]
Josh Achiam, Steven Adler, Sandhini Agarwal, Lama Ahmad, Ilge Akkaya, Florencia Leoni Ale- man, Diogo Almeida, Janko Altenschmidt, Sam Altman, Shyamal Anadkat, et al. Gpt-4 technical report.arXiv preprint arXiv:2303.08774, 2023. 27
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[3]
Alphafold db: Open repository of protein structure predictions
AlphaFold Database Consortium. Alphafold db: Open repository of protein structure predictions. https://alphafold.ebi.ac.uk/, 2024
work page 2024
-
[4]
The claude model family.https://www.anthropic.com/claude, 2024
Anthropic. The claude model family.https://www.anthropic.com/claude, 2024
work page 2024
-
[5]
Anthropic. Claude code: a command-line tool for agentic coding.https://docs.anthropic.c om/en/docs/claude-code, 2025
work page 2025
-
[6]
Liddia: Language-based intelligent drug discovery agent
Reza Averly, Frazier N Baker, Ian A Watson, and Xia Ning. Liddia: Language-based intelligent drug discovery agent. InProceedings of the 2025 Conference on Empirical Methods in Natural Language Processing, pages 12015–12039, 2025
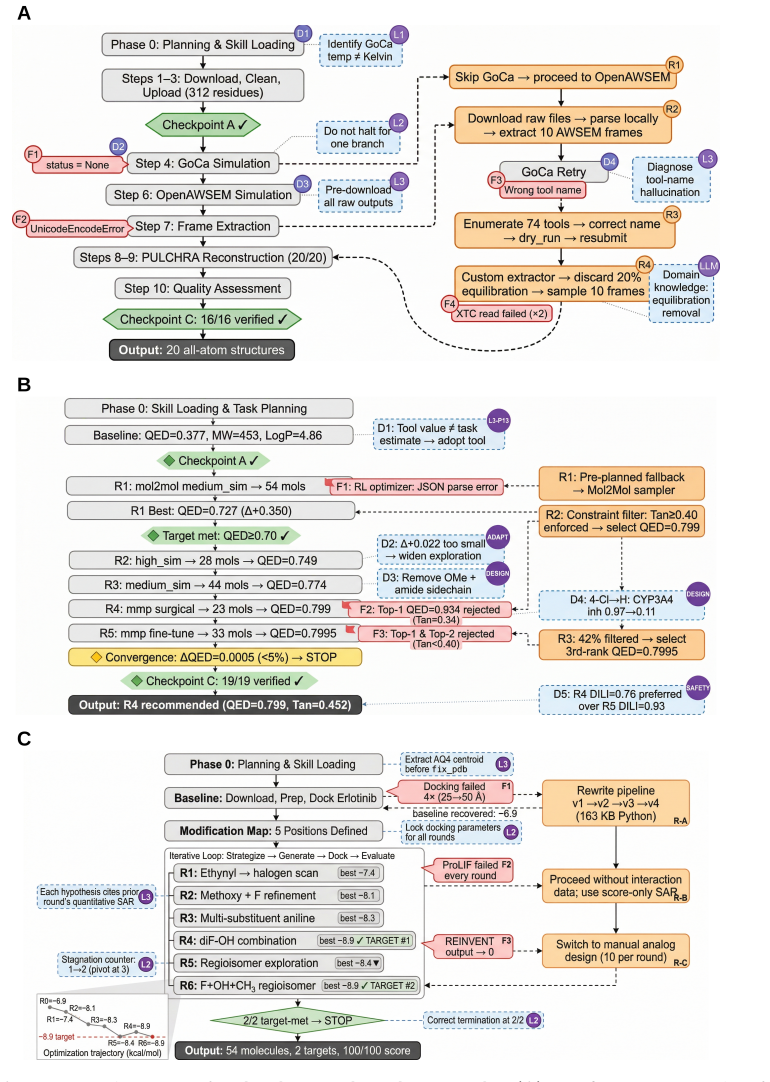
work page 2025
-
[7]
Jinze Bai, Shuai Bai, Yunfei Chu, Zeyu Cui, Kai Dang, Xiaodong Deng, Yang Fan, Wenbin Ge, Yu Han, Fei Huang, et al. Qwen technical report.arXiv preprint arXiv:2309.16609, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[8]
Andreas Bender and Isidro Cortés-Ciriano. Artificial intelligence in drug discovery: what is realistic, what are illusions? part 1: Ways to make an impact, and why we are not there yet. Drug discovery today, 26(2):511–524, 2021
work page 2021
-
[9]
Quantifying the chemical beauty of drugs.Nature Chemistry, 4:90–98, 2012
G Richard Bickerton, Gaia V Paolini, Jérémy Besnard, Sorel Muresan, and Andrew L Hopkins. Quantifying the chemical beauty of drugs.Nature Chemistry, 4:90–98, 2012
work page 2012
-
[10]
Autonomous chemical research with large language models.Nature, 624(7992):570–578, 2023
Daniil A Boiko, Robert MacKnight, Ben Kline, and Gabe Gomes. Autonomous chemical research with large language models.Nature, 624(7992):570–578, 2023
work page 2023
-
[11]
Cédric Bouysset and Sébastien Fiorucci. Prolif: a library to encode molecular interactions as fingerprints.Journal of cheminformatics, 13(1):72, 2021
work page 2021
-
[12]
Evobind: in silico directed evolution of peptide binders with alphafold.bioRxiv, pages 2022–07, 2022
Patrick Bryant and Arne Elofsson. Evobind: in silico directed evolution of peptide binders with alphafold.bioRxiv, pages 2022–07, 2022
work page 2022
-
[13]
Duanhua Cao, Geng Chen, Jiaxin Jiang, Jie Yu, Runze Zhang, Mingan Chen, Wei Zhang, Lifan Chen, Feisheng Zhong, Yingying Zhang, et al. Generic protein–ligand interaction scoring by inte- grating physical prior knowledge and data augmentation modelling.Nature Machine Intelligence, 6(6):688–700, 2024
work page 2024
-
[14]
Mozi: Governed autonomy for drug discovery llm agents.arXiv preprint arXiv:2603.03655, 2026
He Cao, Siyu Liu, Fan Zhang, Zijing Liu, Hao Li, Bin Feng, Shengyuan Bai, Leqing Chen, Kai Xie, and Yu Li. Mozi: Governed autonomy for drug discovery llm agents.arXiv preprint arXiv:2603.03655, 2026
-
[15]
Chembl: The global bioactivity database for drug discovery.https: //www.ebi.ac.uk/chembl/, 2024
ChEMBL Consortium. Chembl: The global bioactivity database for drug discovery.https: //www.ebi.ac.uk/chembl/, 2024
work page 2024
-
[16]
Gabriele Corso, Hannes Stärk, Bowen Jing, Regina Barzilay, and Tommi Jaakkola. Diffdock: Diffusion steps, twists, and turns for molecular docking.arXiv preprint arXiv:2210.01776, 2022
-
[17]
Robust deep learning–based protein sequence design using proteinmpnn.Science, 378(6615):49–56, 2022
Justas Dauparas, Ivan Anishchenko, Nathaniel Bennett, Hua Bai, Robert J Ragotte, Lukas F Milles, Basile IM Wicky, Alexis Courbet, Rob J de Haas, Neville Bethel, et al. Robust deep learning–based protein sequence design using proteinmpnn.Science, 378(6615):49–56, 2022
work page 2022
-
[18]
Pymol: An open-source molecular graphics tool.CCP4 Newsl
Warren L DeLano et al. Pymol: An open-source molecular graphics tool.CCP4 Newsl. protein crystallogr, 40(1):82–92, 2002
work page 2002
-
[19]
Leading ai-driven drug discovery platforms: 2025 landscape and global outlook
Mahendiran Dharmasivam, Busra Kaya, Adedoyin Akinware, Mahan Gholam Azad, and Des R Richardson. Leading ai-driven drug discovery platforms: 2025 landscape and global outlook. Pharmacological Reviews, page 100102, 2025
work page 2025
-
[20]
Ji Ding, Shidi Tang, Zheming Mei, Lingyue Wang, Qinqin Huang, Haifeng Hu, Ming Ling, and Jiansheng Wu. Vina-gpu 2.0: further accelerating autodock vina and its derivatives with graphics processing units.Journal of chemical information and modeling, 63(7):1982–1998, 2023. 28
work page 1982
-
[21]
Peter Eastman, Jason Swails, John D Chodera, Robert T McGibbon, Yutong Zhao, Kyle A Beauchamp, Lee-Ping Wang, Andrew C Simmonett, Matthew P Harrigan, Chaya D Stern, et al. Openmm 7: Rapid development of high performance algorithms for molecular dynamics.PLoS computational biology, 13(7):e1005659, 2017
work page 2017
-
[22]
Jerome Eberhardt, Diogo Santos-Martins, Andreas F Tillack, and Stefano Forli. Autodock vina 1.2. 0: new docking methods, expanded force field, and python bindings.Journal of chemical information and modeling, 61(8):3891–3898, 2021
work page 2021
-
[23]
Shiyang Feng, Runmin Ma, Xiangchao Yan, Yue Fan, Yusong Hu, Songtao Huang, Shuaiyu Zhang, Zongsheng Cao, Tianshuo Peng, Jiakang Yuan, et al. Internagent-1.5: A unified agentic framework for long-horizon autonomous scientific discovery.arXiv preprint arXiv:2602.08990, 2026
-
[24]
Glide: a new approach for rapid, accurate docking and scoring
Richard A Friesner, Jay L Banks, Robert B Murphy, Thomas A Halgren, Jasna J Klicic, Daniel T Mainz, Matthew P Repasky, Eric H Knoll, Mee Shelley, Jason K Perry, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. Journal of medicinal chemistry, 47(7):1739–1749, 2004
work page 2004
-
[25]
Empowering biomedical discovery with ai agents.Cell, 187(22):6125–6151, 2024
Shanghua Gao, Ada Fang, Yepeng Huang, Valentina Giunchiglia, Ayush Noori, Jonathan Richard Schwarz, Yasha Ektefaie, Jovana Kondic, and Marinka Zitnik. Empowering biomedical discovery with ai agents.Cell, 187(22):6125–6151, 2024
work page 2024
-
[26]
Shanghua Gao, Richard Zhu, Zhenglun Kong, Ayush Noori, Xiaorui Su, Curtis Ginder, Theodoros Tsiligkaridis, and Marinka Zitnik. Txagent: an ai agent for therapeutic reasoning across a universe of tools.arXiv preprint arXiv:2503.10970, 2025
-
[27]
Democratising real-world drug discovery through agentic ai.Drug Discovery Today, page 104605, 2026
Jiazhen He, Helen Lai, Lakshidaa Saigiridharan, Gian Marco Ghiandoni, Kinga Jenei, Umur Gokalp, Ajsa Nukovic, Ola Engkvist, Jon Paul Janet, and Samuel Genheden. Democratising real-world drug discovery through agentic ai.Drug Discovery Today, page 104605, 2026
work page 2026
-
[28]
Biomni: A general-purpose biomedical ai agent.biorxiv, 2025
Kexin Huang, Serena Zhang, Hanchen Wang, Yuanhao Qu, Yingzhou Lu, Yusuf Roohani, Ryan Li, Lin Qiu, Gavin Li, Junze Zhang, et al. Biomni: A general-purpose biomedical ai agent.biorxiv, 2025
work page 2025
-
[29]
Illuminating protein space with a programmable generative model.Nature, 623(7989):1070–1078, 2023
John B Ingraham, Max Baranov, Zak Costello, Karl W Barber, Wujie Wang, Ahmed Ismail, VincentFrappier, DanaMLord, ChristopherNg-Thow-Hing, ErikRVanVlack, etal. Illuminating protein space with a programmable generative model.Nature, 623(7989):1070–1078, 2023
work page 2023
-
[30]
Yankai Jiang, Wenjie Lou, Lilong Wang, Zhenyu Tang, Shiyang Feng, Jiaxuan Lu, Haoran Sun, Yaning Pan, Shuang Gu, Haoyang Su, et al. Scp: Accelerating discovery with a global web of autonomous scientific agents.arXiv preprint arXiv:2512.24189, 2025
-
[31]
José Jiménez, Stefan Doerr, Gerard Martínez-Rosell, Alexander S Rose, and Gianni De Fabritiis. Deepsite: protein-binding site predictor using 3d-convolutional neural networks.Bioinformatics, 33(19):3036–3042, 2017
work page 2017
-
[32]
Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
JohnJumper, RichardEvans, AlexanderPritzel, TimGreen, MichaelFigurnov, OlafRonneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
work page 2021
-
[33]
Radoslav Krivák and David Hoksza. P2rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure.Journal of cheminformatics, 10(1):39, 2018
work page 2018
-
[34]
Fpocket: an open source platform for ligand pocket detection.BMC bioinformatics, 10(1):168, 2009
Vincent Le Guilloux, Peter Schmidtke, and Pierre Tuffery. Fpocket: an open source platform for ligand pocket detection.BMC bioinformatics, 10(1):168, 2009
work page 2009
-
[35]
Sarah Lewis, Tim Hempel, José Jiménez-Luna, Michael Gastegger, Yu Xie, Andrew YK Foong, Victor García Satorras, Osama Abdin, Bastiaan S Veeling, Iryna Zaporozhets, et al. Scalable emulation of protein equilibrium ensembles with generative deep learning.Science, 389(6761):eadv9817, 2025. 29
work page 2025
-
[36]
Hao Li, He Cao, Bin Feng, Yanjun Shao, Xiangru Tang, Zhiyuan Yan, Li Yuan, Yonghong Tian, and Yu Li. Beyond chemical qa: Evaluating llm’s chemical reasoning with modular chemical operations.arXiv preprint arXiv:2505.21318, 2025
-
[37]
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
work page 2023
-
[38]
Drugagent: Automating ai-aided drug discovery programming through llm multi-agent collaboration
SizheLiu, YizhouLu, SiyuChen, XiyangHu, JieyuZhao, YingzhouLu, andYueZhao. Drugagent: Automating ai-aided drug discovery programming through llm multi-agent collaboration.arXiv preprint arXiv:2411.15692, 2024
-
[39]
Reinvent 4: Modern ai–driven generative molecule design.Journal of Cheminformatics, 16(1):20, 2024
Hannes H Loeffler, Jiazhen He, Alessandro Tibo, Jon Paul Janet, Alexey Voronov, Lewis H Mervin, and Ola Engkvist. Reinvent 4: Modern ai–driven generative molecule design.Journal of Cheminformatics, 16(1):20, 2024
work page 2024
-
[40]
Wei Lu, Carlos Bueno, Nicholas P Schafer, Joshua Moller, Shikai Jin, Xun Chen, Mingchen Chen, Xinyu Gu, Aram Davtyan, Juan J de Pablo, et al. Openawsem with open3spn2: A fast, flexible, and accessible framework for large-scale coarse-grained biomolecular simulations.PLoS computational biology, 17(2):e1008308, 2021
work page 2021
-
[41]
Bran, Sam Cox, Oliver Schilter, Carlo Baldassari, Andrew D White, and Philippe Schwaller
Andres M. Bran, Sam Cox, Oliver Schilter, Carlo Baldassari, Andrew D White, and Philippe Schwaller. Augmenting large language models with chemistry tools.Nature machine intelligence, 6(5):525–535, 2024
work page 2024
-
[42]
Augmented Language Models: a Survey
Grégoire Mialon, Roberto Dessì, Maria Lomeli, Christoforos Nalmpantis, Ram Pasunuru, Roberta Raileanu, Baptiste Rozière, Timo Schick, Jane Dwivedi-Yu, Asli Celikyilmaz, et al. Augmented language models: a survey.arXiv preprint arXiv:2302.07842, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[43]
OpenClaw Contributors. Openclaw: an open-source framework for building tool-augmented llm agents.https://github.com/openclaw, 2025
work page 2025
-
[44]
Frogent: An end-to-end full-process drug design agent.arXiv preprint arXiv:2508.10760, 2025
Qihua Pan, Dong Xu, Jenna Xinyi Yao, Lijia Ma, Zexuan Zhu, and Junkai Ji. Frogent: An end-to-end full-process drug design agent.arXiv preprint arXiv:2508.10760, 2025
-
[45]
Boltz-2: Towards accurate and efficient binding affinity prediction.BioRxiv, 2025
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, et al. Boltz-2: Towards accurate and efficient binding affinity prediction.BioRxiv, 2025
work page 2025
-
[46]
Pubchem: Open chemistry database.https://pubchem.ncbi.nlm.nih .gov/, 2025
PubChem Consortium. Pubchem: Open chemistry database.https://pubchem.ncbi.nlm.nih .gov/, 2025
work page 2025
-
[47]
RCSB PDB Consortium. Rcsb pdb: Research collaboratory for structural bioinformatics protein data bank.https://www.rcsb.org/, 2024
work page 2024
-
[48]
Rdkit: Open-source cheminformatics software.https://www.rdkit.org/, 2024
RDKit Consortium. Rdkit: Open-source cheminformatics software.https://www.rdkit.org/, 2024
work page 2024
-
[49]
Piotr Rotkiewicz and Jeffrey Skolnick. Fast procedure for reconstruction of full-atom protein models from reduced representations.Journal of computational chemistry, 29(9):1460–1465, 2008
work page 2008
-
[50]
Computational approaches streamlining drug discovery.Nature, 616(7958):673–685, 2023
Anastasiia V Sadybekov and Vsevolod Katritch. Computational approaches streamlining drug discovery.Nature, 616(7958):673–685, 2023
work page 2023
-
[51]
Sebastian Salentin, Sven Schreiber, V Joachim Haupt, Melissa F Adasme, and Michael Schroeder. Plip: fully automated protein–ligand interaction profiler.Nucleic acids research, 43(W1):W443– W447, 2015
work page 2015
-
[52]
Timo Schick, Jane Dwivedi-Yu, Roberto Dessì, Roberta Raileanu, Maria Lomeli, Eric Hambro, Luke Zettlemoyer, Nicola Cancedda, and Thomas Scialom. Toolformer: Language models can teach themselves to use tools.Advances in neural information processing systems, 36:68539– 68551, 2023. 30
work page 2023
-
[53]
Understanding and predicting druggability
Peter Schmidtke and Xavier Barril. Understanding and predicting druggability. a high-throughput method for detection of drug binding sites.Journal of medicinal chemistry, 53(15):5858–5867, 2010
work page 2010
-
[54]
Duxin Sun, Wei Gao, Hongxiang Hu, and Simon Zhou. Why 90% of clinical drug development fails and how to improve it?Acta Pharmaceutica Sinica B, 12(7):3049–3062, 2022
work page 2022
-
[55]
Kyle Swanson, Parker Walther, Jeremy Leitz, Souhrid Mukherjee, Joseph C Wu, Rabindra V Shivnaraine, and James Zou. Admet-ai: a machine learning admet platform for evaluation of large-scale chemical libraries.Bioinformatics, 40(7):btae416, 2024
work page 2024
-
[56]
Chai-1: Decoding the molecular interactions of life
Chai Discovery team, Jacques Boitreaud, Jack Dent, Matthew McPartlon, Joshua Meier, Vinicius Reis, Alex Rogozhonikov, and Kevin Wu. Chai-1: Decoding the molecular interactions of life. BioRxiv, pages 2024–10, 2024
work page 2024
-
[57]
Gemini: A Family of Highly Capable Multimodal Models
Gemini Team, Rohan Anil, Sebastian Borgeaud, Jean-Baptiste Alayrac, Jiahui Yu, Radu Soricut, Johan Schalkwyk, Andrew M Dai, Anja Hauth, Katie Millican, et al. Gemini: A family of highly capable multimodal models. arxiv 2023.arXiv preprint arXiv:2312.11805, 2024
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[58]
Uniprot: The universal protein knowledgebase.https://www.unipro t.org/, 2025
The UniProt Consortium. Uniprot: The universal protein knowledgebase.https://www.unipro t.org/, 2025
work page 2025
-
[59]
Tingzhong Tian, Shuya Li, Ziting Zhang, Lin Chen, Ziheng Zou, Dan Zhao, and Jianyang Zeng. Benchmarking compound activity prediction for real-world drug discovery applications.Commu- nications Chemistry, 7(1):127, 2024
work page 2024
-
[60]
LLaMA: Open and Efficient Foundation Language Models
Hugo Touvron, Thibaut Lavril, Gautier Izacard, Xavier Martinet, Marie-Anne Lachaux, Timothée Lacroix, Baptiste Rozière, Naman Goyal, Eric Hambro, Faisal Azhar, et al. Llama: Open and efficient foundation language models.arXiv preprint arXiv:2302.13971, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[61]
Mario S Valdés-Tresanco, Mario E Valdés-Tresanco, Pedro A Valiente, and Ernesto Moreno. gmx_mmpbsa: a new tool to perform end-state free energy calculations with gromacs.Journal of chemical theory and computation, 17(10):6281–6291, 2021
work page 2021
-
[62]
Jessica Vamathevan, Dominic Clark, Paul Czodrowski, Ian Dunham, Edgardo Ferran, George Lee, Bin Li, Anant Madabhushi, Parantu Shah, Michaela Spitzer, et al. Applications of machine learning in drug discovery and development.Nature reviews Drug discovery, 18(6):463–477, 2019
work page 2019
-
[63]
Ivana Vichentijevikj, Kostadin Mishev, and Monika Simjanoska Misheva. Prompt-to-pill: Multi- agent drug discovery and clinical simulation pipeline.Bioinformatics Advances, 6(1):vbaf323, 2026
work page 2026
-
[64]
Luis J Walter, Patrick K Quoika, and Martin Zacharias. Structure-based protein assembly simu- lations including various binding sites and conformations.Journal of Chemical Information and Modeling, 64(8):3465–3476, 2024
work page 2024
-
[65]
Scientific discovery in the age of artificial intelligence.Nature, 620(7972):47–60, 2023
Hanchen Wang, Tianfan Fu, Yuanqi Du, Wenhao Gao, Kexin Huang, Ziming Liu, Payal Chandak, Shengchao Liu, Peter Van Katwyk, Andreea Deac, et al. Scientific discovery in the age of artificial intelligence.Nature, 620(7972):47–60, 2023
work page 2023
-
[66]
Olivier J Wouters, Martin McKee, and Jeroen Luyten. Estimated research and development investment needed to bring a new medicine to market, 2009-2018.Jama, 323(9):844–853, 2020
work page 2009
-
[67]
The hdock server for integrated protein–protein docking.Nature protocols, 15(5):1829–1852, 2020
Yumeng Yan, Huanyu Tao, Jiahua He, and Sheng-You Huang. The hdock server for integrated protein–protein docking.Nature protocols, 15(5):1829–1852, 2020
work page 2020
-
[68]
React: Synergizing reasoning and acting in language models
Shunyu Yao, Jeffrey Zhao, Dian Yu, Nan Du, Izhak Shafran, Karthik R Narasimhan, and Yuan Cao. React: Synergizing reasoning and acting in language models. InThe eleventh international conference on learning representations, 2022
work page 2022
-
[69]
Xujun Zhang, Odin Zhang, Chao Shen, Wanglin Qu, Shicheng Chen, Hanqun Cao, Yu Kang, Zhe Wang, Ercheng Wang, Jintu Zhang, et al. Efficient and accurate large library ligand docking with karmadock.Nature Computational Science, 3(9):789–804, 2023. 31
work page 2023
-
[70]
Activity cliff prediction: Dataset and benchmark.arXiv preprint arXiv:2302.07541, 2023
Ziqiao Zhang, Bangyi Zhao, Ailin Xie, Yatao Bian, and Shuigeng Zhou. Activity cliff prediction: Dataset and benchmark.arXiv preprint arXiv:2302.07541, 2023
-
[71]
Jie Zhu, Jingxiang Wang, Xin Wang, Mingjing Gao, Bingbing Guo, Miaomiao Gao, Jiarui Liu, Yanqiu Yu, Liang Wang, Weikaixin Kong, et al. Prediction of drug efficacy from transcriptional profiles with deep learning.Nature biotechnology, 39(11):1444–1452, 2021. Data A vailability Both the MolBench dataset (CSV format) and associated evaluation code can be acc...
work page 2021
-
[72]
Here we derive this bound analytically
Predicting the optimization ceiling from scaffold topology A central finding of this study is that the triazolo-benzodiazepine scaffold imposes a hard upper bound on the achievable QED score. Here we derive this bound analytically. QED is defined as a weighted geometric mean of eight component desirability functionsdi (ref. [9]): QED= exp P8 i=1 wi lnd i ...
-
[73]
Tanimoto budget exhaustion as a convergence diagnostic In our main Results we noted that the qualification rate—the fraction of generated molecules satisfying the Tanimoto≥0.40 constraint—declined from 100% (R1–R2) to 57.6% (R5). We propose that this declining rate constitutes a generalizable convergence diagnostic that we term “Tanimoto budget exhaustion...
-
[74]
Phase transitions versus gradual improvement in property optimization Not all QED components improved gradually. The structural alerts (ALERTS) desirability exhibited a discontinuous phase transition: 0.241 at R0 to 0.842 at R1, with no further change in R2–R5. This occurred because the starting molecule’s butyl ester triggered two Brenk structural alerts...
-
[75]
propose 3–5 modified molecules with chemical ratio- nale
The interpretability–efficiency trade-off in generative design The evaluation question instructed the agent to “propose 3–5 modified molecules with chemical ratio- nale.” Instead, the agent employed REINVENT4 batch generation to produce 23–54 candidates per round and selected the best by QED ranking. This substitution raises a fundamental question about A...
-
[76]
Systematic blind spot detection in multi-objective optimization The agent’s failure to detect the AMES mutagenicity deterioration (+180%, from 0.165 to 0.462) de- spite tracking over 13 ADMET endpoints illustrates a general vulnerability of attention-based monitor- ing. The agent explicitly tracked CYP3A4, hERG and DILI at each round—all of which improved...
-
[77]
Cost-effectiveness and practical stopping rules Thediminishingreturnspattern(Fig.7H)hasdirectimplicationsforcomputationalresourceallocation. Assuming roughly equal tool-call costs per round, and measuring against the R0-to-R4 improvement (+0.4216) since R4 is the recommended molecule, R1 delivers 83.0%, R1–R2 deliver 88.1%, R1–R3 deliver 94.0%, and R1–R4 ...
-
[78]
Multi-round iterative optimization as an emergent agent capability The E2E-Q3 task required the AI agent to execute an iterative closed-loop optimization cycle— Strategize, Generate, Dock, Evaluate—across up to 15 rounds, with autonomous decision-making at each round boundary. This represents a fundamentally different challenge from single-step computa- t...
-
[79]
Long-range planning, self-repair, and emergent medicinal chemistry knowledge The agent autonomously authored four pipeline versions (v1–v4, totaling 163 KB of Python), pro- gressively diagnosing and recovering from crashes: v1 failed due to NumPy/RDKit incompatibility, v2 succeeded through R1 but crashed on an f-string bug, v3 resumed from R2 with pre-pro...
-
[80]
Agent versus REINVENT: complementary collaboration rather than competition The 3:3 tie in round winners and the non-significant pooled comparison (p= 0.104) mask a deeper complementarity. REINVENT excelled at creative molecular recombination: it serendipitously dis- covered methoxy shortening in R1 (not hypothesized by the agent), generated the F+OH+CH3 m...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.