CaliPPer: quantifying, predicting and improving AI model performance for binding prediction
Pith reviewed 2026-06-27 20:24 UTC · model grok-4.3
The pith
CaliPPer uses sample-to-domain distances and Bayesian recalibration to predict and improve binding model performance on new data without labels.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
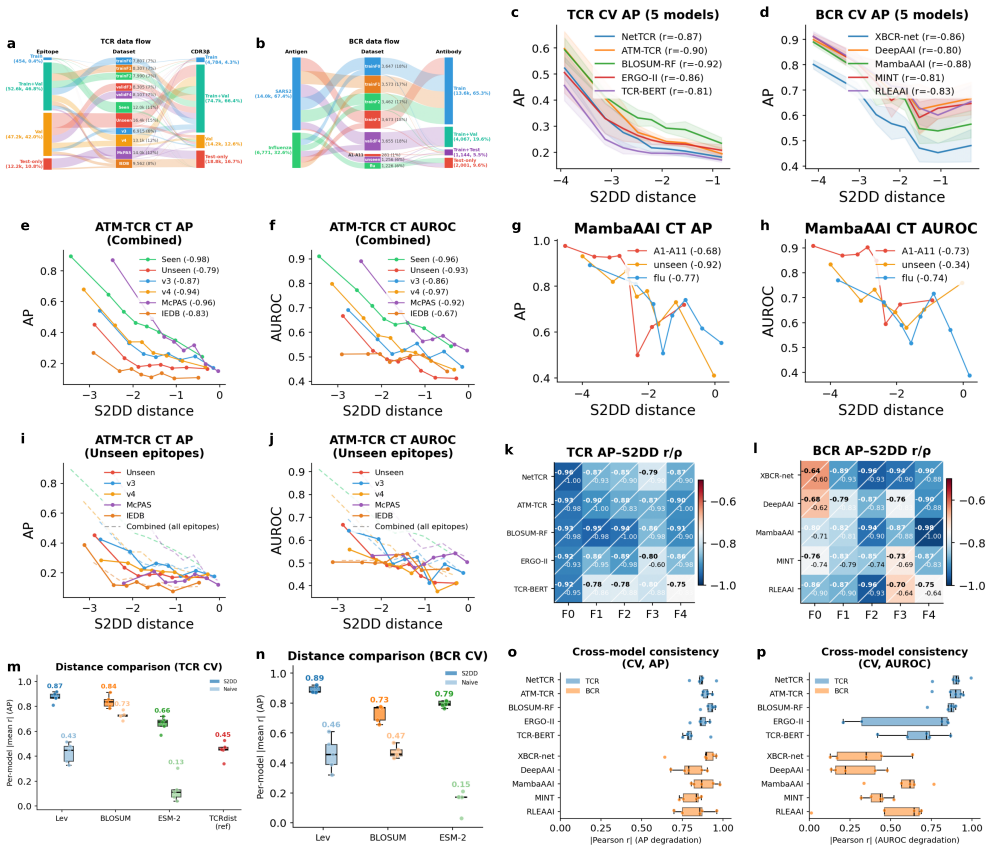
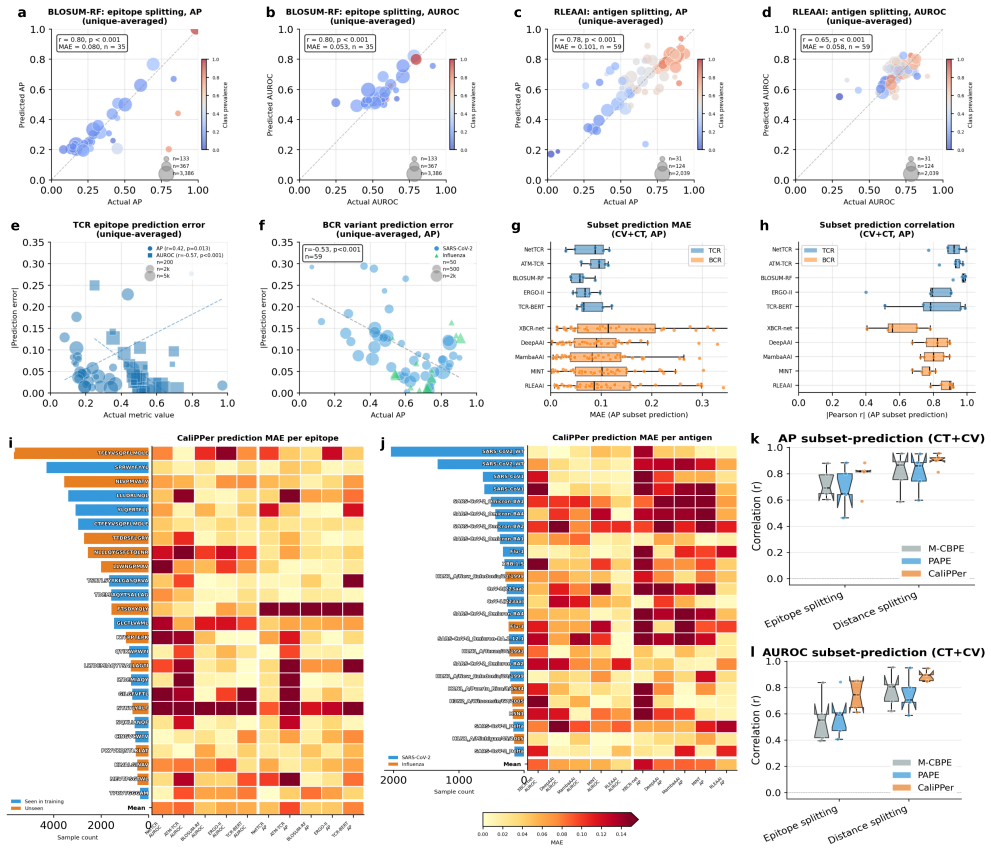
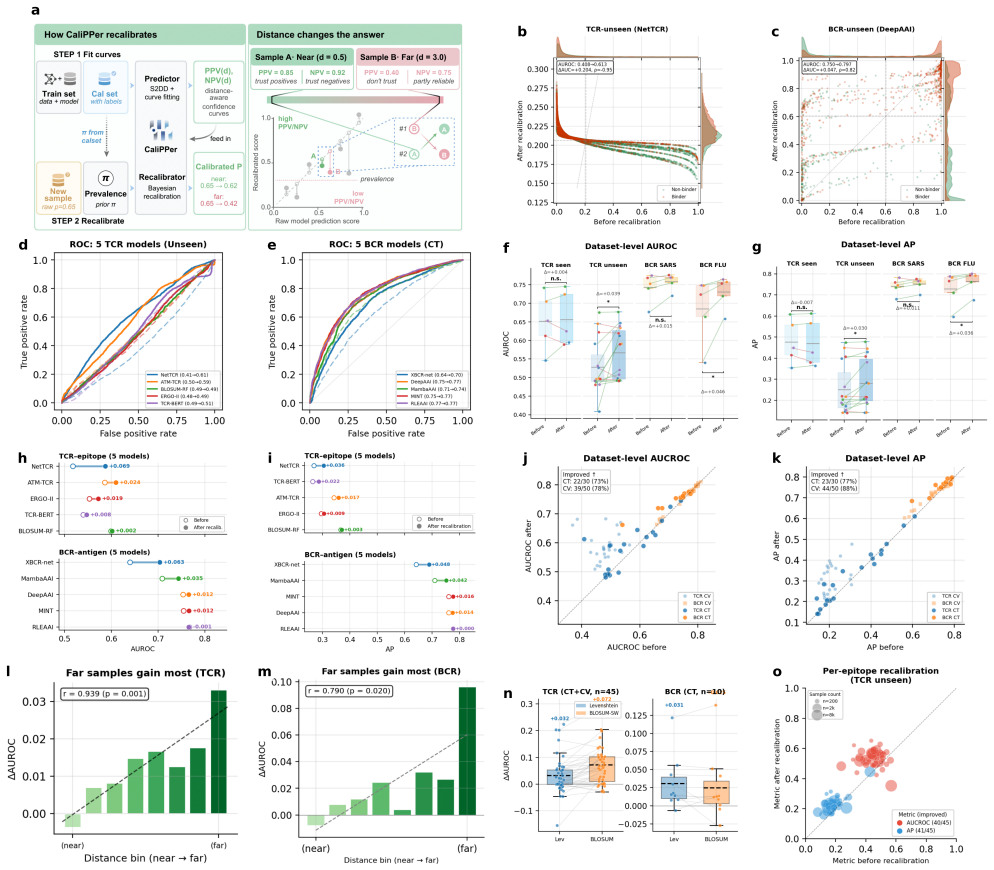
CaliPPer pairs a multi-chain Sample-to-Domain Distance (S2DD) with distance-aware Bayesian recalibration to provide label-free generalisability scores, aggregate performance predictions with low error, and improved per-sample predictions for binding models, demonstrated by high correlations and AUROC gains on unseen data across ten models and two domains.
What carries the argument
Multi-chain Sample-to-Domain Distance (S2DD) combined with distance-aware Bayesian recalibration, which quantifies deviation from training data and adjusts model outputs accordingly.
If this is right
- Distance-performance correlations reach |r|=0.80–0.92 across tested models.
- Performance metrics like AUROC are predicted with mean absolute errors of 0.008–0.070.
- AUROC improves by up to +0.20 on unseen epitopes and variants.
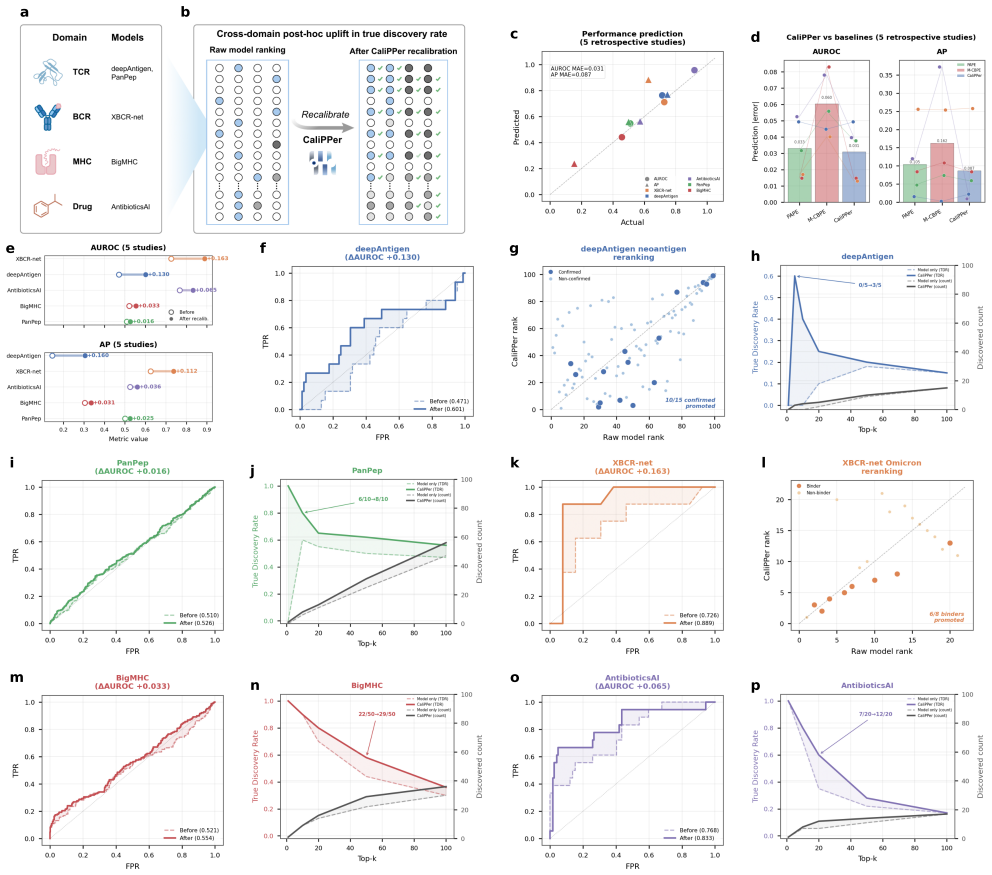
- True discovery rates increase in all five retrospectively analyzed published studies.
- The method requires no retraining and no target labels.
Where Pith is reading between the lines
- If the distance metric captures relevant domain shifts, the framework could apply to other machine learning tasks involving distribution shift.
- Per-sample confidence scores may help prioritize which predictions to validate experimentally first.
- Combining this with model training might yield even better generalisation.
- Similar approaches could address performance unpredictability in other scientific prediction domains like protein design.
Load-bearing premise
That the Sample-to-Domain Distance reliably signals how performance will drop on new data for the range of models and domains tested.
What would settle it
Observing low or inconsistent distance-performance correlations (below |r|=0.6) or high prediction errors (above 0.1 MAE) on a new set of binding models and datasets would falsify the central claims.
Figures


read the original abstract
Binding prediction models accelerate therapeutic antibody and TCR discovery, but their performance on new datasets is unpredictable, often leading to low discovery rates. Density-ratio methods (PAPE, M-CBPE) provide label-free performance estimation for binary classification, but their assumptions and aggregate-only outputs limit binding prediction on neoepitopes, antigen variants and chemical scaffolds. Here we present CaliPPer (Calibration and Prediction of Performance), a post-hoc framework pairing a multi-chain Sample-to-Domain Distance (S2DD) with distance-aware Bayesian recalibration, operating at three resolutions: generalisability score, aggregate performance prediction, and per-sample confidence. Across ten models, eight architectures and two immune-receptor domains, CaliPPer attains distance--performance correlations $|r|=0.80\text{--}0.92$, predicts AUROC/AP/F1 with mean absolute errors $0.008\text{--}0.070$, and improves AUROC by up to $+0.20$ on unseen epitopes/variants. Applied retrospectively to five published TCR, BCR, MHC--peptide and small-molecule studies, CaliPPer raises true discovery rates in all five (e.g.\ $0/5 \to 3/5$ confirmed neoantigens), providing a triage layer between computational prediction and experimental validation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces CaliPPer, a post-hoc framework that combines a multi-chain Sample-to-Domain Distance (S2DD) metric with distance-aware Bayesian recalibration to quantify generalisability, predict aggregate performance (AUROC/AP/F1), and provide per-sample confidence scores for binding prediction models. Across ten models, eight architectures, and two immune-receptor domains, it reports distance-performance correlations of |r|=0.80–0.92, mean absolute prediction errors of 0.008–0.070, AUROC gains up to +0.20 on unseen epitopes/variants, and retrospective improvements in true discovery rates when applied to five published TCR/BCR/MHC-peptide/small-molecule studies.
Significance. If the empirical results hold under rigorous validation, CaliPPer could supply a practical label-free triage layer for assessing and enhancing binding predictors in therapeutic discovery pipelines. The multi-resolution outputs and retrospective application to real studies are concrete strengths; the absence of free parameters and the focus on transfer across domains without retraining are also positive features. However, the provided text supplies insufficient experimental detail to confirm that the reported correlations and improvements are not artifacts of data leakage or post-hoc selection.
major comments (3)
- [Abstract] Abstract: the central claims rest on numerical results (correlations |r|=0.80–0.92, MAEs 0.008–0.070, AUROC gains +0.20) but the text supplies no information on experimental design, train/test splits, baseline methods, error-bar reporting, or pre-specification of the ten models and eight architectures; without these the support for the label-free transfer claim cannot be evaluated.
- [Methods] Methods (S2DD definition): the multi-chain Sample-to-Domain Distance must be shown to be independent of the downstream performance labels; if S2DD incorporates any quantity derived from the same binding data used for AUROC/AP evaluation, the reported correlations would be circular by construction.
- [Results] Results (retrospective application): the claim that CaliPPer raises true discovery rates in all five published studies (e.g., 0/5 → 3/5) requires explicit reporting of how the distance threshold and recalibration were chosen without access to the held-out experimental labels; otherwise the improvement may reflect hindsight selection.
minor comments (2)
- [Abstract] Abstract: specify whether the reported |r| values are Pearson or Spearman correlations and list the exact number of models per domain.
- [Introduction] Notation: the acronym S2DD should be expanded on first use and its mathematical definition given before any correlation plots.
Simulated Author's Rebuttal
We thank the referee for the constructive comments highlighting the need for greater transparency in experimental details and methodological independence. We address each major comment below and will revise the manuscript to incorporate additional clarifications and explicit statements.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claims rest on numerical results (correlations |r|=0.80–0.92, MAEs 0.008–0.070, AUROC gains +0.20) but the text supplies no information on experimental design, train/test splits, baseline methods, error-bar reporting, or pre-specification of the ten models and eight architectures; without these the support for the label-free transfer claim cannot be evaluated.
Authors: We agree that the abstract would benefit from additional context on the experimental scope. In the revised manuscript we will expand the abstract with a concise statement on the datasets (immune-receptor domains), the ten models spanning eight architectures, and the cross-domain evaluation protocol. Full details on train/test splits, baseline comparisons, error bars, and pre-specification are already present in the Methods and Supplementary Information; we will add explicit cross-references from the abstract to these sections. revision: yes
-
Referee: [Methods] Methods (S2DD definition): the multi-chain Sample-to-Domain Distance must be shown to be independent of the downstream performance labels; if S2DD incorporates any quantity derived from the same binding data used for AUROC/AP evaluation, the reported correlations would be circular by construction.
Authors: S2DD is computed solely from input features (sequence embeddings or structural representations) between query samples and reference domain samples; no binding labels, affinity values, or performance metrics enter the distance calculation at any stage. We will insert a new subsection in Methods containing the formal definition, a short proof of label independence, and pseudocode that explicitly excludes any label-derived quantities. revision: yes
-
Referee: [Results] Results (retrospective application): the claim that CaliPPer raises true discovery rates in all five published studies (e.g., 0/5 → 3/5) requires explicit reporting of how the distance threshold and recalibration were chosen without access to the held-out experimental labels; otherwise the improvement may reflect hindsight selection.
Authors: The distance thresholds and Bayesian recalibration parameters were fixed in advance using only source-domain S2DD statistics (e.g., median plus one standard deviation) and a pre-defined heuristic; these rules were applied to the five target studies without reference to their experimental labels. We will add a dedicated paragraph and supplementary table documenting the exact protocol, the source-only data used for parameter setting, and the corresponding code to demonstrate the label-free procedure. revision: yes
Circularity Check
No significant circularity; empirical validation stands independent of inputs
full rationale
The paper presents CaliPPer as an empirical post-hoc framework whose performance (distance-performance correlations, MAE on AUROC/AP/F1 predictions, AUROC gains) is measured on held-out epitopes/variants and retrospective published studies. No derivation chain is claimed that reduces a 'prediction' to a fitted parameter by construction, nor does any equation define S2DD or the Bayesian recalibration in terms of the target performance metrics. The central claims rest on external validation across ten models and five studies rather than self-referential definitions or self-citation load-bearing steps. Absent any quoted reduction of output to input, the derivation is self-contained.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Zhou, L.et al.General scales unlock ai evaluation with explanatory and predictive power.Nature652, 58–67, DOI: 10.1038/s41586-026-10303-2 (2026)
-
[2]
Wong, F.et al.Discovery of a structural class of antibiotics with explainable deep learning.Nature626, 177–185, DOI: 10.1038/s41586-023-06887-8 (2024)
-
[3]
Biol.4, 1060, DOI: 10.1038/s42003-021-02610-3 (2021)
Montemurro, A.et al.Nettcr-2.0 enables accurate prediction of tcr-peptide binding by using paired tcr α and β sequence data.Commun. Biol.4, 1060, DOI: 10.1038/s42003-021-02610-3 (2021)
-
[4]
Cai, M., Bang, S., Zhang, P. & Lee, H. Atm-tcr: Tcr-epitope binding affinity prediction using a multi-head self-attention model.Front. Immunol.13, 893247, DOI: 10.3389/fimmu.2022.893247 (2022). 7/27
-
[5]
Lou, H.et al.Deep learning-based rapid generation of broadly reactive antibodies against sars-cov-2 and its omicron variant.Cell Res.33, 80–82, DOI: 10.1038/s41422-022-00727-6 (2023)
-
[6]
immunology13, 1014256, DOI: 10.3389/fimmu.2022.1014256 (2022)
Grazioli, F.et al.On tcr binding predictors failing to generalize to unseen peptides.Front. immunology13, 1014256, DOI: 10.3389/fimmu.2022.1014256 (2022)
-
[7]
ImmunoInformatics9, 100024, DOI: 10.1016/j.immuno.2023.100024 (2023)
Meysman, P.et al.Benchmarking solutions to the T-cell receptor epitope prediction problem: Immrep22 workshop report. ImmunoInformatics9, 100024, DOI: 10.1016/j.immuno.2023.100024 (2023)
-
[8]
Methods23, 248–259, DOI: 10.1038/s41592-025-02910-0 (2026)
Lu, Y .et al.Assessment of computational methods in predicting tcr–epitope binding recognition.Nat. Methods23, 248–259, DOI: 10.1038/s41592-025-02910-0 (2026)
-
[9]
Zhang, J.et al.Predicting unseen antibodies’ neutralizability via adaptive graph neural networks.Nat. Mach. Intell.4, 964–976, DOI: 10.1038/s42256-022-00553-w (2022)
-
[10]
Hu, J., Zhou, Y ., Zhang, W.-Y . & Zhou, X.-G. RLEAAI: improving antibody-antigen interaction prediction using protein language model and sequence order information.Briefings Bioinforma.26, bbaf238, DOI: 10.1093/bib/bbaf238 (2025)
-
[11]
& Weinberger, K
Guo, C., Pleiss, G., Sun, Y . & Weinberger, K. Q. On calibration of modern neural networks. InInternational conference on machine learning, 1321–1330 (PMLR, 2017)
2017
-
[12]
Neural Inf
Ovadia, Y .et al.Can you trust your model’s uncertainty? Evaluating predictive uncertainty under dataset shift.Adv. Neural Inf. Process. Syst.32(2019)
2019
-
[13]
Wang, N.et al.Deep peptide recognition profiling decodes tcr specificity and enables disease-associated antigen discovery. Nat. Biotechnol.1–11, DOI: 10.1038/s41587-026-03128-x (2026). Advance online publication
-
[14]
& Perrakis, N
Białek, J., Kivim¨aki, J., Kuberski, W. & Perrakis, N. Estimating model performance under covariate shift without labels. InAdvances in Neural Information Processing Systems, vol. 38, 161084–161115 (Curran Associates, Inc., 2025)
2025
-
[15]
Kivim¨aki, J., Nurminen, J. K., Białek, J. & Kuberski, W. Confidence-based estimators for predictive performance in model monitoring.J. Artif. Intell. Res.82, 209–240, DOI: 10.1613/jair.1.16709 (2025)
-
[16]
Dash, P.et al.Quantifiable predictive features define epitope-specific T cell receptor repertoires.Nature547, 89–93, DOI: 10.1038/nature22383 (2017)
-
[17]
I.et al.Binary codes capable of correcting deletions, insertions, and reversals.Sov
Levenshtein, V . I.et al.Binary codes capable of correcting deletions, insertions, and reversals.Sov. Phys. Doklady10, 707–710 (1966)
1966
-
[18]
Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks.Proc. Natl. Acad. Sci.89, 10915–10919, DOI: 10.1073/pnas.89.22.10915 (1992)
-
[19]
Lin, Z.et al.Evolutionary-scale prediction of atomic-level protein structure with a language model.Science379, 1123–1130, DOI: 10.1126/science.ade2574 (2023)
-
[20]
Springer, I., Besser, H., Tickotsky-Moskovitz, N., Dvorkin, S. & Louzoun, Y . Prediction of specific TCR-peptide binding from large dictionaries of TCR-peptide pairs.Front. Immunol.11, 1803, DOI: 10.3389/fimmu.2020.01803 (2020)
-
[21]
E.et al.TCR-BERT: learning the grammar of T-cell receptors for flexible antigen-binding analyses
Wu, K. E.et al.TCR-BERT: learning the grammar of T-cell receptors for flexible antigen-binding analyses. InProceedings of the 18th Machine Learning in Computational Biology meeting, vol. 240, 194–229 (PMLR, 2024)
2024
-
[22]
Ullanat, V ., Jing, B., Sledzieski, S. & Berger, B. Learning the language of protein-protein interactions.Nat. Commun.17, 1199, DOI: 10.1038/s41467-025-67971-3 (2026)
-
[23]
Liu, X., Fu, H., Yang, Y . & Zhang, J. Bio-inspired Mamba for antibody-antigen interaction prediction.Biomolecules15, 764, DOI: 10.3390/biom15060764 (2025)
-
[24]
Pham, M.-D. N.et al.epitcr: a highly sensitive predictor for tcr–peptide binding.Bioinformatics39, btad284, DOI: 10.1093/bioinformatics/btad284 (2023)
-
[25]
Commun.16, 5171, DOI: 10.1038/s41467-025-60461-6 (2025)
Que, J.et al.Identifying t cell antigen at the atomic level with graph convolutional network.Nat. Commun.16, 5171, DOI: 10.1038/s41467-025-60461-6 (2025)
-
[26]
Gao, Y .et al.Pan-peptide meta learning for t-cell receptor–antigen binding recognition.Nat. Mach. Intell.5, 236–249, DOI: 10.1038/s42256-023-00619-3 (2023)
-
[27]
Albert, B. A.et al.Deep neural networks predict class i major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity.Nat. machine intelligence5, 861–872, DOI: 10.1038/s42256-023-00694-6 (2023)
-
[28]
Tickotsky, N., Sagiv, T., Prilusky, J., Shifrut, E. & Friedman, N. McPAS-TCR: a manually curated catalogue of pathology- associated T cell receptor sequences.Bioinformatics33, 2924–2929, DOI: 10.1093/bioinformatics/btx286 (2017)
-
[29]
Vita, R.et al.The Immune Epitope Database (IEDB): 2018 update.Nucleic acids research47, D339–D343, DOI: 10.1093/nar/gky1006 (2019)
-
[30]
Immunol.16, 1488851, DOI: 10.3389/fimmu.2025.1488851 (2025)
Nolan, S.et al.A large-scale database of T-cell receptor beta (TCR β) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2.Front. Immunol.16, 1488851, DOI: 10.3389/fimmu.2025.1488851 (2025)
-
[31]
V ., Grazioli, F., Machart, P., M¨osch, A
Castorina, L. V ., Grazioli, F., Machart, P., M¨osch, A. & Errica, F. Assessing the generalization capabilities of TCR binding predictors via peptide distance analysis.PLoS One20, e0324011, DOI: 10.1371/journal.pone.0324011 (2025)
-
[32]
Jumper, J.et al.Highly accurate protein structure prediction with AlphaFold.Nature596, 583–589, DOI: 10.1038/ s41586-021-03819-2 (2021)
2021
-
[33]
Platt, J. C. Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods. In Advances in Large Margin Classifiers, 61–74 (MIT Press, 1999)
1999
-
[34]
Youden, W. J. Index for rating diagnostic tests.Cancer3, 32–35, DOI: 10.1002/1097-0142(1950)3:1 ⟨32:: AID-CNCR2820030106⟩3.0.CO;2-3 (1950)
-
[35]
Bagaev, D. V .et al.Vdjdb in 2019: database extension, new analysis infrastructure and a t-cell receptor motif compendium. Nucleic acids research48, D1057–D1062, DOI: 10.1093/nar/gkz874 (2020). 8/27 a b c 1 Sample-to-Domain Distance (S2DD) i Chain-to-chain distance ii Chain-to-Sample weighting iii Sample-to-Domain Reduction O U T P U T S2DD distance d=D (...
-
[36]
Deep learning-based prediction of the T cell receptor–antigen binding specificity
Tianshi Lu, Ze Zhang, James Zhu, et al. Deep learning-based prediction of the T cell receptor–antigen binding specificity. Nature Machine Intelligence, 3:864–875, 2021
2021
-
[37]
Can we predict T cell specificity with digital biology and machine learning?Nature Reviews Immunology, 23:511–521, 2023
Dan Hudson, Ricardo A Fernandes, Mark Basham, Graham Ogg, and Hashem Koohy. Can we predict T cell specificity with digital biology and machine learning?Nature Reviews Immunology, 23:511–521, 2023
2023
-
[38]
CoV-AbDab: the coronavirus antibody database.Bioinformatics, 37(5):734–735, 2021
Matthew I J Raybould, Aleksandr Kovaltsuk, Claire Marks, and Charlotte M Deane. CoV-AbDab: the coronavirus antibody database.Bioinformatics, 37(5):734–735, 2021
2021
-
[39]
Vaccine-induced antibodies that neutralize group 1 and group 2 influenza A viruses.Cell, 166(3):609–623, 2016
M Gordon Joyce, Adam K Wheatley, Paul V Thomas, et al. Vaccine-induced antibodies that neutralize group 1 and group 2 influenza A viruses.Cell, 166(3):609–623, 2016
2016
-
[40]
Cross-lineage protection by human antibodies binding the influenza B hemagglutinin.Nature Communications, 10:324, 2019
Yi Liu, Hyon-Xhi Tan, Marios Koutsakos, et al. Cross-lineage protection by human antibodies binding the influenza B hemagglutinin.Nature Communications, 10:324, 2019
2019
-
[41]
Immune history profoundly affects broadly protective B cell responses to influenza.Science Translational Medicine, 7(316):316ra192, 2015
Sarah F Andrews, Yunping Huang, Kaval Kaur, et al. Immune history profoundly affects broadly protective B cell responses to influenza.Science Translational Medicine, 7(316):316ra192, 2015
2015
-
[42]
Influenza virus vaccination elicits poorly adapted B cell responses in elderly individuals.Cell Host & Microbe, 25(3):357–366, 2019
Carole Henry, Nai-Ying Zheng, Min Huang, et al. Influenza virus vaccination elicits poorly adapted B cell responses in elderly individuals.Cell Host & Microbe, 25(3):357–366, 2019
2019
-
[43]
Preexisting immunity shapes distinct antibody landscapes after influenza virus infection and vaccination in humans.Science Translational Medicine, 12(573):eabd3601, 2020
Haley L Dugan, Jenna J Guthmiller, Philip Arevalo, et al. Preexisting immunity shapes distinct antibody landscapes after influenza virus infection and vaccination in humans.Science Translational Medicine, 12(573):eabd3601, 2020
2020
-
[44]
Flow cytometry reveals that H5N1 vaccination elicits cross-reactive stem-directed antibodies from multiple Ig heavy-chain lineages.Journal of Virology, 88(8):4047–4057, 2014
James RR Whittle, Adam K Wheatley, Lan Wu, et al. Flow cytometry reveals that H5N1 vaccination elicits cross-reactive stem-directed antibodies from multiple Ig heavy-chain lineages.Journal of Virology, 88(8):4047–4057, 2014
2014
-
[45]
Carole Henry, Anna-Karin E Palm, Henry A Utset, et al. Monoclonal antibody responses after recombinant hemagglutinin vaccine versus subunit inactivated influenza virus vaccine: a comparative study.Journal of Virology, 93(21):e01150–19, 2019
2019
-
[46]
Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection.Journal of Experimental Medicine, 208(1):181–193, 2011
Jens Wrammert, Dimitrios Koutsonanos, Gui-Mei Li, et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection.Journal of Experimental Medicine, 208(1):181–193, 2011
2009
-
[47]
Preferential induction of cross-group influenza A hemagglutinin stem-specific memory B cells after H7N9 immunization in humans.Science Immunology, 2(13):eaan2676, 2017
Sarah F Andrews, M Gordon Joyce, Michael J Chambers, et al. Preferential induction of cross-group influenza A hemagglutinin stem-specific memory B cells after H7N9 immunization in humans.Science Immunology, 2(13):eaan2676, 2017
2017
-
[48]
Crystal Sao-Fong Cheung, Alexander Fruehwirth, Philipp Carl Georg Paparoditis, et al. Identification and structure of a multidonor class of head-directed influenza-neutralizing antibodies reveal the mechanism for its recurrent elicitation.Cell Reports, 32(9):108088, 2020
2020
-
[49]
Focused antibody response to influenza linked to antigenic drift.Journal of Clinical Investigation, 125(7):2631–2645, 2015
Kuan-Ying A Huang, Pramila Rijal, Lisa Schimanski, et al. Focused antibody response to influenza linked to antigenic drift.Journal of Clinical Investigation, 125(7):2631–2645, 2015
2015
-
[50]
Broadly neutralizing antibodies target a haemagglutinin anchor epitope.Nature, 602:314–320, 2022
Jenna J Guthmiller, Julianna Han, Henry A Utset, et al. Broadly neutralizing antibodies target a haemagglutinin anchor epitope.Nature, 602:314–320, 2022
2022
-
[51]
First exposure to the pandemic H1N1 virus induced broadly neutralizing antibodies targeting hemagglutinin head epitopes.Science Translational Medicine, 13(596):eabg4535, 2021
Jenna J Guthmiller, Julianna Han, Lei Li, et al. First exposure to the pandemic H1N1 virus induced broadly neutralizing antibodies targeting hemagglutinin head epitopes.Science Translational Medicine, 13(596):eabg4535, 2021
2021
-
[52]
Xueyong Zhu, Julianna Han, Weina Sun, et al. Influenza chimeric hemagglutinin structures in complex with broadly protec- tive antibodies to the stem and trimer interface.Proceedings of the National Academy of Sciences, 119(21):e2200821119, 2022
2022
-
[53]
Mosaic nanoparticle display of diverse influenza virus hemagglutinins elicits broad B cell responses.Nature Immunology, 20(3):362–372, 2019
Masaru Kanekiyo, M Gordon Joyce, Rebecca A Gillespie, et al. Mosaic nanoparticle display of diverse influenza virus hemagglutinins elicits broad B cell responses.Nature Immunology, 20(3):362–372, 2019
2019
-
[54]
Highly conserved protective epitopes on influenza B viruses.Science, 337(6100):1343–1348, 2012
Cyrille Dreyfus, Nick S Laursen, Ted Kwaks, et al. Highly conserved protective epitopes on influenza B viruses.Science, 337(6100):1343–1348, 2012
2012
-
[55]
CD8+ T cells of Healthy Donor 1–4
Nicole L Kallewaard, Davide Corti, Patrick J Collins, et al. Structure and function analysis of an antibody recognizing all influenza A subtypes.Cell, 166(3):596–608, 2016. Data Availability TCR–epitope binding data were assembled from VDJdb35 and 10x Genomics CD8+ T-cell dextramer datasets (UMI-denoised; four healthy donors, “CD8+ T cells of Healthy Dono...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.