DeepBD: A Grounded Agentic Workflow for Variant Prioritization and Diagnosis of Genetic Birth Defects
Pith reviewed 2026-06-25 21:28 UTC · model grok-4.3
The pith
A grounded agentic workflow called DeepBD ranks genetic variants for birth defects by combining rule evidence, mechanistic context and specialist modules into layered prioritization.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
DeepBD organizes variant prioritization and diagnostic interpretation into LLM-assisted case structuring, a pretrained evidence engine that learns patient-specific scores from structured rule evidence, sequence and variant-effect representations and phenotype-conditioned biological context, specialist evidence modules for tool-based refinement, and a grounded diagnostic review layer, achieving the reported recall rates on held-out solved cases while outperforming the listed baselines.
What carries the argument
The grounded agentic workflow that separates evidence integration from tool-based refinement and LLM-assisted diagnostic review.
If this is right
- Rule evidence, mechanistic context and specialist refinement provide complementary signals for variant ranking.
- The workflow outperforms standalone Exomiser, DeepRare and prompted LLM reranking when applied to Exomiser-derived top-20 candidate lists.
- Ablation and overlap analyses confirm that each layer contributes distinct information to the final prioritization.
Where Pith is reading between the lines
- The separation of evidence integration, refinement and review layers may transfer to variant interpretation tasks in other Mendelian disorders.
- Adding direct functional assay data or real-time clinical notes could further improve the evidence engine scores.
- Clinical adoption would require prospective testing on diverse populations to check whether the internal benchmark performance holds outside the development cohort.
Load-bearing premise
The internal held-out benchmark from the authors' 18,622-case cohort is representative of real-world clinical cases and free of selection bias or data leakage.
What would settle it
Evaluation of the same workflow on an independent external cohort of solved genetic birth defect cases in which recall at rank 1, 3, 5 or 10 does not exceed the performance of Exomiser or the other baselines.
Figures

read the original abstract
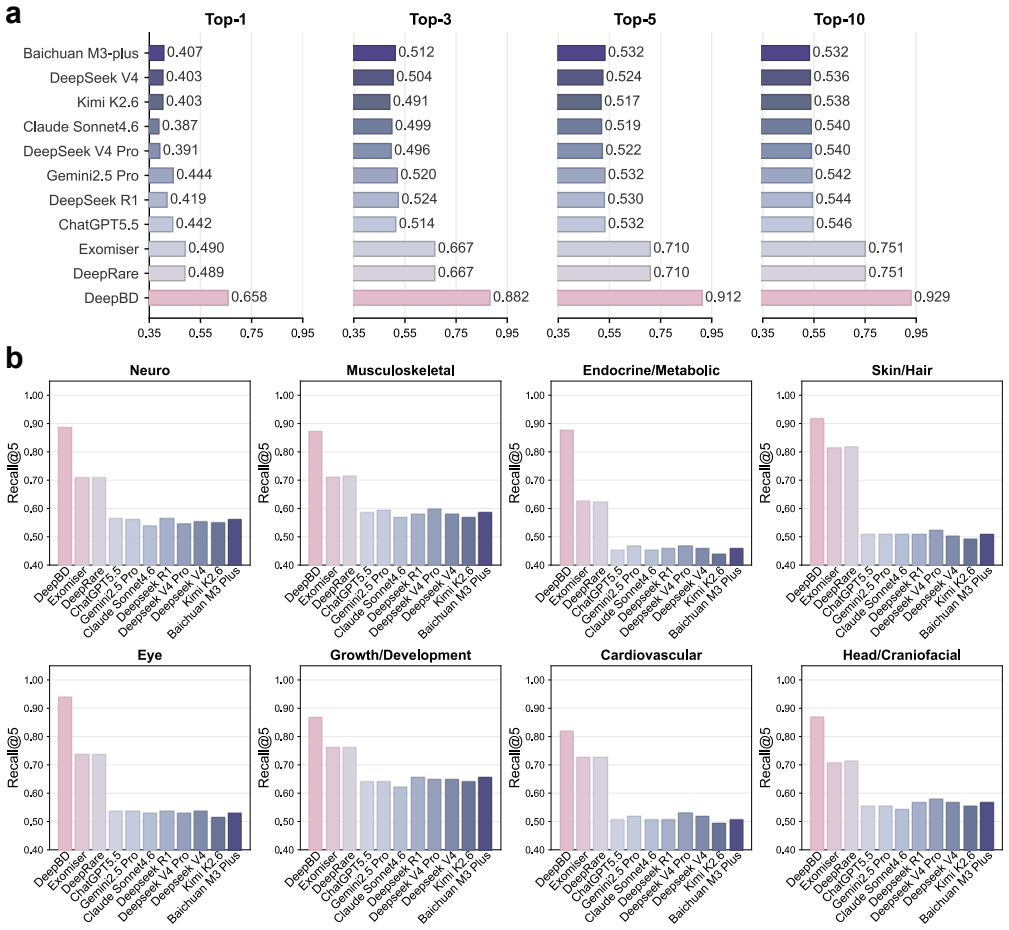
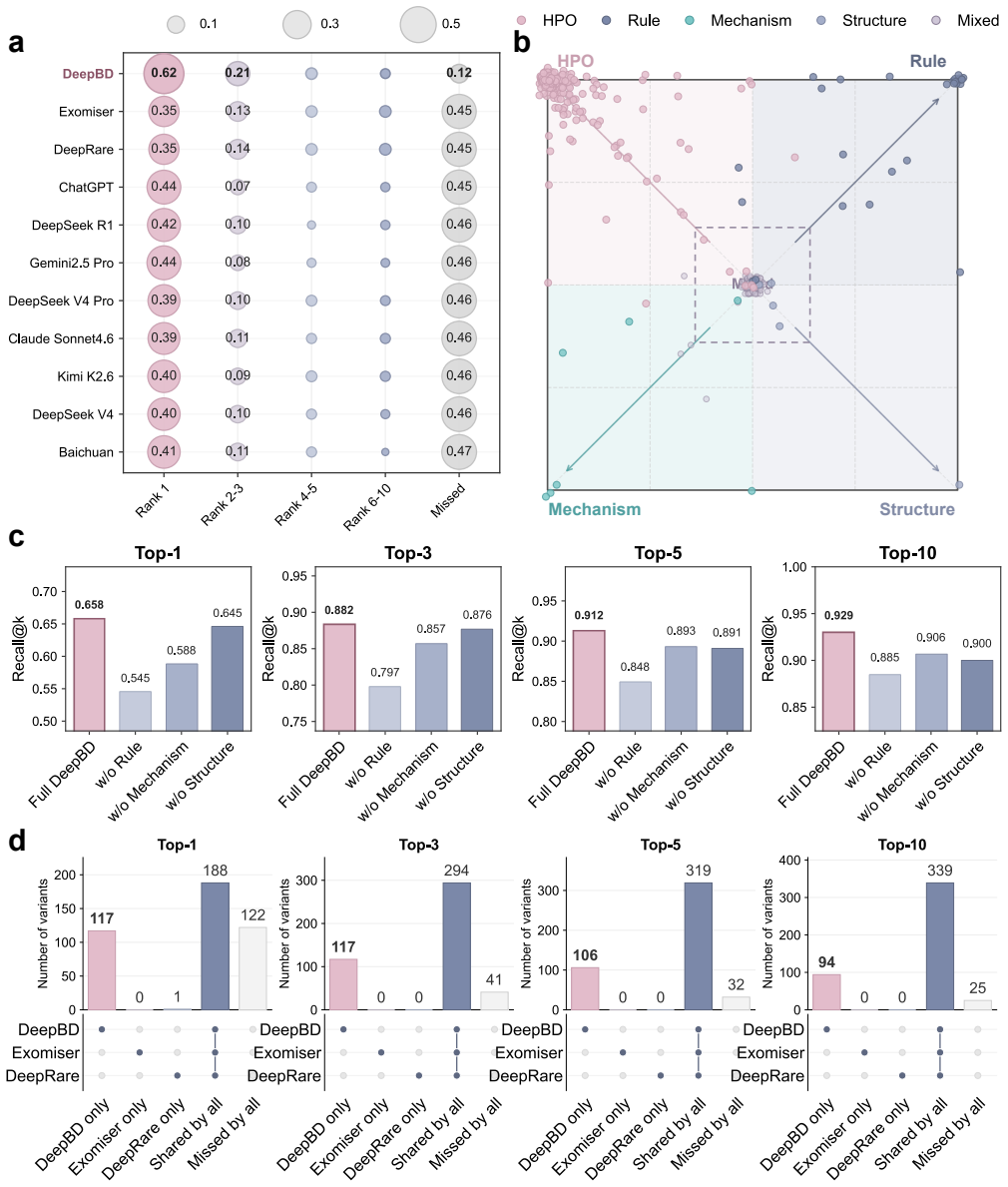
Birth defects are a major cause of fetal loss, neonatal morbidity and long-term disability. In the subset with suspected genetic etiologies, exome and genome sequencing have moved many cases from variant detection to post-sequencing interpretation: clinicians must rank patient-specific candidate variants under incomplete fetal or infant phenotypes and heterogeneous evidence from population genetics, variant-effect prediction, gene-disease validity, phenotype ontologies, cellular and pathway context, protein structure and clinical literature. We present DeepBD, a grounded agentic workflow for variant prioritization and diagnostic interpretation of genetic birth defects. DeepBD organizes the workflow into LLM-assisted case structuring, a pretrained evidence engine, specialist evidence modules and a grounded diagnostic review layer. The evidence engine learns patient-specific variant scores from structured rule evidence, sequence and variant-effect representations and phenotype-conditioned biological context, whereas specialist modules and the agentic layer provide tool-based refinement, candidate-pool review and diagnosis-oriented synthesis from ranked candidates. Developed using an in-house fetal and infant cohort comprising 18,622 cases, DeepBD achieved Recall@1/3/5/10 of 0.658/0.882/0.912/0.929 on an internal held-out solved-case benchmark, outperforming standalone Exomiser, DeepRare and prompted LLM reranking baselines evaluated on Exomiser-derived top-20 candidate variants. Ablation and overlap analyses show that rule evidence, mechanistic context, and specialist refinement provide complementary signals. These findings support a grounded agentic workflow that separates evidence integration, tool-based refinement, and LLM-assisted diagnostic review for retrospective variant prioritization in genetic birth defects.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents DeepBD, a grounded agentic workflow for variant prioritization and diagnostic interpretation of genetic birth defects. It combines LLM-assisted case structuring, a pretrained evidence engine that learns patient-specific variant scores from structured rule evidence, sequence/variant-effect representations and phenotype-conditioned biological context, specialist evidence modules for tool-based refinement, and a grounded diagnostic review layer. Developed on an in-house cohort of 18,622 fetal/infant cases, DeepBD reports Recall@1/3/5/10 of 0.658/0.882/0.912/0.929 on an internal held-out solved-case benchmark, outperforming standalone Exomiser, DeepRare and prompted LLM reranking baselines (evaluated on Exomiser top-20 candidates). Ablation and overlap analyses indicate complementary signals from rule evidence, mechanistic context and specialist refinement.
Significance. If the reported performance generalizes, the separation of evidence integration from tool-based refinement and LLM-assisted synthesis offers a structured approach to handling heterogeneous data sources in birth-defect variant interpretation. The ablations demonstrating non-redundant contributions from different modules are a constructive element of the work.
major comments (3)
- [Evaluation / Results] Evaluation section (and Methods): The held-out benchmark is drawn exclusively from the same 18,622-case in-house development cohort, with no reported patient-level de-duplication, variant-overlap checks between train and test, phenotype stratification of the split, or exclusion criteria. This directly undermines the independence required for the superiority claim over baselines.
- [Results / Methods] Results and Methods: All performance numbers (including the Recall@K values and comparisons to Exomiser/DeepRare/LLM baselines) are reported only on internal held-out data; no external validation cohort, no error bars or confidence intervals, and no details on the evidence-engine training procedure or hyper-parameter selection are provided.
- [Abstract / Results] Abstract and Results: The central claim that DeepBD 'outperforms' the baselines rests on a benchmark whose construction risks circularity, because the evidence engine learns scores from the identical cohort distribution used for the held-out test; without independent data this does not establish generalizability.
minor comments (2)
- [Methods] Clarify in the Methods whether the 'pretrained' evidence engine uses any data sources external to the 18,622-case cohort or is trained entirely in-house.
- [Methods / Supplementary] Add patient-level statistics (e.g., number of unique probands, phenotype distributions) for both the development and held-out sets to allow assessment of selection bias.
Simulated Author's Rebuttal
We thank the referee for their thorough and constructive review. We address each major comment below, indicating planned revisions to strengthen the description of the evaluation methodology while remaining faithful to the work performed.
read point-by-point responses
-
Referee: [Evaluation / Results] Evaluation section (and Methods): The held-out benchmark is drawn exclusively from the same 18,622-case in-house development cohort, with no reported patient-level de-duplication, variant-overlap checks between train and test, phenotype stratification of the split, or exclusion criteria. This directly undermines the independence required for the superiority claim over baselines.
Authors: We agree that explicit documentation of the split construction is required. In the revised manuscript we will expand the Methods section to detail the patient-level splitting procedure, including de-duplication steps, variant-overlap checks between training and test partitions, phenotype stratification, and exclusion criteria applied to the 18,622-case cohort. These additions will clarify the degree of independence achieved. revision: yes
-
Referee: [Results / Methods] Results and Methods: All performance numbers (including the Recall@K values and comparisons to Exomiser/DeepRare/LLM baselines) are reported only on internal held-out data; no external validation cohort, no error bars or confidence intervals, and no details on the evidence-engine training procedure or hyper-parameter selection are provided.
Authors: We will revise the Results and Methods sections to report error bars or confidence intervals on the Recall@K metrics, and to provide a full description of the evidence-engine training procedure together with the hyper-parameter selection protocol. A new limitations subsection will explicitly state that all reported results are internal to the development cohort. revision: yes
-
Referee: [Abstract / Results] Abstract and Results: The central claim that DeepBD 'outperforms' the baselines rests on a benchmark whose construction risks circularity, because the evidence engine learns scores from the identical cohort distribution used for the held-out test; without independent data this does not establish generalizability.
Authors: We will revise the Abstract and Results sections to qualify all performance claims as applying to an internal held-out benchmark drawn from the same development cohort. We will also add explicit discussion of the shared distributional characteristics and the consequent limits on claims of generalizability, thereby addressing the circularity concern by narrowing the scope of the stated conclusions. revision: yes
- We do not currently possess an independent external validation cohort for this specialized fetal/infant birth-defect application and therefore cannot supply external performance numbers without new data collection.
Circularity Check
Internal held-out benchmark from same 18,622-case development cohort makes reported recalls fitted quantities rather than independent predictions
specific steps
-
fitted input called prediction
[Abstract]
"Developed using an in-house fetal and infant cohort comprising 18,622 cases, DeepBD achieved Recall@1/3/5/10 of 0.658/0.882/0.912/0.929 on an internal held-out solved-case benchmark, outperforming standalone Exomiser, DeepRare and prompted LLM reranking baselines evaluated on Exomiser-derived top-20 candidate variants."
The evidence engine learns patient-specific variant scores from the 18,622-case cohort; the quoted recalls are then measured on held-out cases from that same cohort. The performance numbers are therefore quantities obtained by fitting to the authors' data distribution and evaluating within it, rather than independent predictions on external data.
full rationale
The paper's central empirical claim is the Recall@1/3/5/10 performance on an internal held-out solved-case benchmark drawn from the identical in-house cohort used to develop the evidence engine. This reduces the reported numbers to performance on data the model was fitted to, matching the fitted_input_called_prediction pattern. No external cohort or independent validation set is described in the provided text, and the baselines are also evaluated on the same internal candidates. The workflow description (LLM structuring, specialist modules, agentic review) does not itself reduce to a self-definition or self-citation chain, so the circularity is partial and confined to the performance claim.
Axiom & Free-Parameter Ledger
free parameters (1)
- evidence engine model parameters
axioms (1)
- domain assumption The held-out split from the in-house cohort is unbiased and representative of external clinical cases
Reference graph
Works this paper leans on
-
[1]
Congenital disorders
World Health Organization. Congenital disorders. https://www.who.int/news-room/fact-sheets/detail/birth-defects (2023). Accessed 18 June 2026. 9/16
2023
-
[2]
Data and statistics on birth defects
Centers for Disease Control and Prevention. Data and statistics on birth defects. https://www.cdc.gov/birth-defects/ data-research/facts-stats/index.html (2026). Accessed 18 June 2026
2026
-
[3]
About congenital anomalies
Eunice Kennedy Shriver National Institute of Child Health and Human Development. About congenital anomalies. https://www.nichd.nih.gov/health/topics/factsheets/congenital-anomalies (2024). Accessed 18 June 2026
2024
-
[4]
T.et al.Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies.Am
Miller, D. T.et al.Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies.Am. J. Hum. Genet.86, 749–764 (2010)
2010
-
[5]
G., Leach, N
Monaghan, K. G., Leach, N. T., Pekarek, D., Prasad, P. & Rose, N. C. The use of fetal exome sequencing in prenatal diagnosis: a points to consider document of the American College of Medical Genetics and Genomics.Genet. Med.22, 675–680 (2020)
2020
-
[6]
V ora, N. L. & Norton, M. E. Prenatal exome and genome sequencing for fetal structural abnormalities.Am. J. Obstet. Gynecol.228, 140–149 (2023)
2023
-
[7]
Large-scale discovery of novel genetic causes of developmental disorders
Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature519, 223–228 (2015)
2015
-
[8]
F.et al.Prevalence and architecture of de novo mutations in developmental disorders.Nature542, 433–438 (2017)
McRae, J. F.et al.Prevalence and architecture of de novo mutations in developmental disorders.Nature542, 433–438 (2017)
2017
-
[9]
Lord, J.et al.Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study.The Lancet393, 747–757 (2019)
2019
-
[10]
The Lancet393, 758–767 (2019)
Petrovski, S.et al.Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. The Lancet393, 758–767 (2019)
2019
-
[11]
Fu, F.et al.Application of exome sequencing for prenatal diagnosis of fetal structural anomalies: clinical experience and lessons learned from a cohort of 1618 fetuses.Genome Med.14, 123 (2022)
2022
-
[12]
& Chitty, L
Mellis, R., Oprych, K., Scotchman, E., Hill, M. & Chitty, L. S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: a systematic review and meta-analysis.Prenat. Diagn.42, 662–685 (2022)
2022
-
[13]
H.et al.Diagnostic yield of pediatric and prenatal exome sequencing in a diverse population.npj Genomic Med.8, 34 (2023)
Wojcik, M. H.et al.Diagnostic yield of pediatric and prenatal exome sequencing in a diverse population.npj Genomic Med.8, 34 (2023)
2023
-
[14]
N.et al.The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease.Am
Robinson, P. N.et al.The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease.Am. J. Hum. Genet.83, 610–615 (2008). 15.Köhler, S.et al.The Human Phenotype Ontology in 2021.Nucleic Acids Res.49, D1207–D1217 (2021)
2008
-
[15]
A.et al.The Monarch Initiative in 2019: an integrative data and analytic platform connecting phenotypes to genotypes across species.Nucleic Acids Res.48, D704–D715 (2020)
Shefchek, K. A.et al.The Monarch Initiative in 2019: an integrative data and analytic platform connecting phenotypes to genotypes across species.Nucleic Acids Res.48, D704–D715 (2020)
2019
-
[16]
H., Buske, O
Fujiwara, T., Yamamoto, Y ., Kim, J. H., Buske, O. J. & Takagi, T. PubCaseFinder: a case-report-based, phenotype-driven differential-diagnosis system for rare diseases.Am. J. Hum. Genet.103, 389–399 (2018)
2018
-
[17]
Javed, A., Agrawal, S. & Ng, P. C. Phen-gen: combining phenotype and genotype to analyze rare disorders.Nat. Methods 11, 935–937 (2014)
2014
-
[18]
Protoc.10, 2004–2015 (2015)
Smedley, D.et al.Next-generation diagnostics and disease-gene discovery with the Exomiser.Nat. Protoc.10, 2004–2015 (2015)
2004
-
[19]
D., Ma, X
Li, Q., Zhao, K., Bustamante, C. D., Ma, X. & Wong, W. H. Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis.Genet. Med.21, 2126–2134 (2019)
2019
-
[20]
N.et al.Interpretable clinical genomics with a likelihood ratio paradigm.Am
Robinson, P. N.et al.Interpretable clinical genomics with a likelihood ratio paradigm.Am. J. Hum. Genet.107, 403–417 (2020)
2020
-
[21]
Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function.Nucleic Acids Res.31, 3812–3814 (2003)
2003
-
[22]
A.et al.A method and server for predicting damaging missense mutations.Nat
Adzhubei, I. A.et al.A method and server for predicting damaging missense mutations.Nat. Methods7, 248–249 (2010)
2010
-
[23]
Genet.46, 310–315 (2014)
Kircher, M.et al.A general framework for estimating the relative pathogenicity of human genetic variants.Nat. Genet.46, 310–315 (2014)
2014
-
[24]
M., Rödelsperger, C., Schuelke, M
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations.Nat. Methods7, 575–576 (2010). 10/16
2010
-
[25]
M.et al.REVEL: an ensemble method for predicting the pathogenicity of rare missense variants.Am
Ioannidis, N. M.et al.REVEL: an ensemble method for predicting the pathogenicity of rare missense variants.Am. J. Hum. Genet.99, 877–885 (2016)
2016
-
[26]
Genet.50, 1161–1170 (2018)
Sundaram, L.et al.Predicting the clinical impact of human mutation with deep neural networks.Nat. Genet.50, 1161–1170 (2018). 28.Jaganathan, K.et al.Predicting splicing from primary sequence with deep learning.Cell176, 535–548.e24 (2019)
2018
-
[27]
Cheng, J.et al.Accurate proteome-wide missense variant effect prediction with AlphaMissense.Science381, eadg7492 (2023)
2023
-
[28]
Med.17, 405–424 (2015)
Richards, S.et al.Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the ACMG and the AMP.Genet. Med.17, 405–424 (2015)
2015
-
[29]
T.et al.Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the Clinical Genome Resource.Genet
Strande, N. T.et al.Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the Clinical Genome Resource.Genet. Med.19, 896–906 (2017)
2017
-
[30]
J.et al.ClinVar: public archive of interpretations of clinically relevant variants.Nucleic Acids Res.44, D862–D868 (2016)
Landrum, M. J.et al.ClinVar: public archive of interpretations of clinically relevant variants.Nucleic Acids Res.44, D862–D868 (2016)
2016
-
[31]
D.et al.The Human Gene Mutation Database (HGMD): optimizing its use in a clinical diagnostic or research setting.Hum
Stenson, P. D.et al.The Human Gene Mutation Database (HGMD): optimizing its use in a clinical diagnostic or research setting.Hum. Genet.139, 1197–1207 (2020)
2020
-
[32]
S., Bocchini, C
Amberger, J. S., Bocchini, C. A., Scott, A. F. & Hamosh, A. OMIM.org: leveraging knowledge across phenotype-gene relationships.Nucleic Acids Res.47, D1038–D1043 (2019)
2019
-
[33]
N., Gkoutos, G
Boudellioua, I., Kulmanov, M., Schofield, P. N., Gkoutos, G. V . & Hoehndorf, R. DeepPVP: phenotype-based prioritization of causative variants using deep learning.BMC Bioinformatics20, 65 (2019)
2019
-
[34]
C.et al.Deep structured learning for variant prioritization in Mendelian diseases.Nat
Danzi, M. C.et al.Deep structured learning for variant prioritization in Mendelian diseases.Nat. Commun.14, 4167 (2023)
2023
-
[35]
Mao, D.et al.AI-MARRVEL: a knowledge-driven AI system for diagnosing Mendelian disorders.NEJM AI1, AIoa2300009 (2024)
2024
-
[36]
Alsentzer, E.et al.Few shot learning for phenotype-driven diagnosis of patients with rare genetic diseases.npj Digit. Med. 8, 380 (2025). 39.Singhal, K.et al.Large language models encode clinical knowledge.Nature620, 172–180 (2023). 40.Tu, T.et al.Towards conversational diagnostic artificial intelligence.Nature642, 442–450 (2025). 41.Zhao, W.et al.An agen...
2025
-
[37]
A Versatile AI Agent for Rare Disease Diagnosis and Risk Gene Prioritization
Liu, T.et al.A versatile AI agent for rare disease diagnosis and risk gene prioritization. Preprint at https://arxiv.org/abs/ 2605.06226 (2026)
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[38]
Preprint at https://doi.org/10.64898/2026.04.02.26349929 (2026)
Meng, M., Liu, L., Du, Q.et al.Berrylyzer: an efficient, traceable, and lightweight intelligent agentic system for prenatal genetic diagnosis. Preprint at https://doi.org/10.64898/2026.04.02.26349929 (2026)
-
[39]
J.et al.The mutational constraint spectrum quantified from variation in 141,456 humans.Nature581, 434–443 (2020)
Karczewski, K. J.et al.The mutational constraint spectrum quantified from variation in 141,456 humans.Nature581, 434–443 (2020)
2020
-
[40]
46.Veli ˇckovi´c, P.et al.Graph Attention Networks
Nguyen, E.et al.Sequence modeling and design from molecular to genome scale with Evo.Science386, eado9336 (2024). 46.Veli ˇckovi´c, P.et al.Graph Attention Networks. InInternational Conference on Learning Representations(2018)
2024
-
[41]
The GTEx Consortium atlas of genetic regulatory effects across human tissues.Science369, 1318–1330 (2020)
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues.Science369, 1318–1330 (2020). 48.Milacic, M.et al.The Reactome pathway knowledgebase 2024.Nucleic Acids Res.52, D672–D678 (2024). 49.Du, J.et al.Gene2vec: distributed representation of genes based on co-expression.BMC Genomics20, 82 (2019). 50.Jumper, J.et al.Hig...
2020
-
[42]
Varadi, M.et al.AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences.Nucleic Acids Res.52, D368–D375 (2024)
2024
-
[43]
J.et al.RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite.PLoS ONE6, e20161 (2011)
Fleishman, S. J.et al.RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite.PLoS ONE6, e20161 (2011)
2011
-
[44]
F.et al.The Rosetta all-atom energy function for macromolecular modeling and design.J
Alford, R. F.et al.The Rosetta all-atom energy function for macromolecular modeling and design.J. Chem. Theory Comput.13, 3031–3048 (2017). 11/16 54.Lek, M.et al.Analysis of protein-coding genetic variation in 60,706 humans.Nature536, 285–291 (2016). 55.The 1000 Genomes Project Consortium. A global reference for human genetic variation.Nature526, 68–74 (2...
2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.