Accelerating Molecular Dynamics Simulations using Fast Ewald Summation with Prolates
Pith reviewed 2026-05-22 14:48 UTC · model grok-4.3
The pith
Prolate spheroidal wave functions shrink the Fourier grid in Ewald summation and accelerate electrostatic forces in molecular dynamics by a factor of three.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
ESP achieves a more compact Fourier representation of the far-field Coulomb potential by expanding the smoothed charge density in prolate spheroidal wave functions rather than Fourier modes; the resulting grid can be made smaller while the truncation error stays below a prescribed tolerance, thereby lowering the arithmetic and communication work of the particle-mesh stage without introducing additional approximation errors beyond those already present in conventional PME or PPPM.
What carries the argument
Ewald summation with prolate spheroidal wave functions (ESP), which replaces the standard trigonometric basis with a PSWF basis chosen to concentrate energy in a smaller number of modes.
If this is right
- Electrostatic force evaluation becomes three times faster at typical production tolerances and up to ten times faster at 10^{-5} tolerance.
- Overall molecular-dynamics throughput rises by a factor of 2.5 on roughly one thousand cores and by a factor of five at high accuracy.
- Strong scaling improves because the smaller grids reduce the amount of all-to-all communication per time step.
- The accelerated kernels can be used as direct replacements inside existing LAMMPS and GROMACS input decks without changing the physical model.
Where Pith is reading between the lines
- The same PSWF compression could be applied to other mesh-based long-range kernels such as gravitational or dispersion forces.
- Lower grid sizes would also reduce peak memory footprint, enabling larger system sizes on a given machine.
- Because the method is already integrated into two major packages, it offers an immediate route to lower energy consumption per nanosecond of simulated time.
- Future extensions might combine the reduced grid with real-space cutoffs or machine-learned short-range potentials to push the overall scaling even closer to linear.
Load-bearing premise
The prolate spheroidal wave function basis reaches the target accuracy with far fewer grid points than a trigonometric basis and does so without new stability problems or extra tuning steps.
What would settle it
Measure the actual force error and wall-clock time when the same trajectory is run once with ESP and once with the baseline PME or PPPM implementation at identical tolerance; if the error exceeds tolerance or the observed speedup falls below roughly 2x on a few hundred cores, the central performance claim is refuted.
Figures




read the original abstract
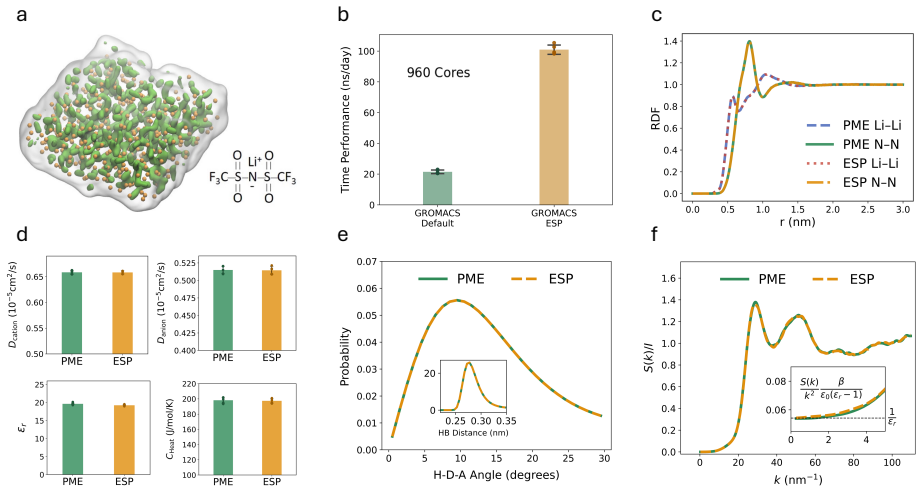
The evaluation of long-range Coulomb interactions is a significant cost in molecular dynamics (MD), even when using Particle Mesh Ewald (PME) or Particle-Particle-Particle-Mesh (PPPM) methods, which rely on Ewald splitting and the fast Fourier transform to achieve near-linear scaling. We introduce ESP -- Ewald summation with prolate spheroidal wave functions (PSWFs) -- which leads to a more efficient Fourier representation and a reduction in the required grid size, global communication, and particle-grid operations, without loss of accuracy. We have integrated the ESP method into two widely-used open-source MD packages, LAMMPS and GROMACS, enabling rapid comparison and adoption. Relative to PME/PPPM baselines at error tolerances $10^{-3}$ to $10^{-4}$, ESP gives roughly a $3$-fold acceleration of electrostatic interactions, and a $2.5$-fold speed-up in the MD simulation when using about $10^3$ compute cores. At high accuracy ($10^{-5}$), these increase to $10$-fold for the far-field electrostatics and $5$-fold for MD simulation. Furthermore, we show that the accelerated codes have improved strong scaling with core count, and validate them in realistic long-time biological and material simulations. ESP thus offers a practical, drop-in path to reduce the time-to-solution and energy footprint of MD workflows.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces ESP (Ewald summation with prolate spheroidal wave functions), which replaces standard Fourier representations in PME/PPPM with a PSWF basis to reduce required grid sizes, global communication, and particle-grid operations in molecular dynamics. The central claims are 3-fold acceleration of electrostatic interactions (rising to 10-fold at 10^{-5} tolerance) and 2.5- to 5-fold overall MD speedups on ~10^3 cores relative to LAMMPS/GROMACS baselines, achieved without loss of accuracy, with validation on realistic biological and materials systems plus improved strong scaling.
Significance. If the accuracy and grid-reduction claims hold, the work offers a practical route to lower time-to-solution and energy costs for long-range electrostatics in production MD codes. Direct integration into LAMMPS and GROMACS plus timing comparisons against established PME/PPPM baselines provide a concrete basis for assessing deployability; reproducible speedups at scale would be a notable contribution to computational molecular science.
major comments (3)
- [Numerical validation] Numerical validation section: RMS force and energy errors are reported only at the final chosen grid sizes for ESP; a controlled side-by-side comparison of the minimal grid dimensions required by ESP versus standard PME/PPPM to meet identical tolerances (10^{-3} to 10^{-5}) is absent. This comparison is load-bearing for the speedup attribution, as the claimed reductions in communication and particle-grid work rest on substantially smaller grids.
- [Method description] Method and error analysis: The simultaneous control of Ewald splitting error, Gaussian screening parameter, and PSWF truncation/projection error under periodic boundaries is not quantified with explicit bounds or sensitivity tests. Any mismatch could introduce approximation errors absent from PME/PPPM; the abstract's 'without loss of accuracy' claim therefore requires a direct demonstration that target tolerances are preserved at the reduced grids.
- [Performance and scaling] Performance tables and scaling results: The reported 2.5- to 5-fold MD speedups at ~10^3 cores should include a breakdown isolating time in particle-grid assignment, FFT, and MPI communication for ESP versus baselines. Without this, it is difficult to confirm that the gains derive from the smaller grids rather than implementation details.
minor comments (2)
- [Abstract] Abstract: 'about 10^3 compute cores' should be replaced by the exact core counts and node configurations used in the timing benchmarks for reproducibility.
- [Figures] Figures: Timing plots should include variability measures (e.g., standard deviation over repeated runs) to account for parallel performance fluctuations.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments on our manuscript. We address each major point below and have revised the manuscript to incorporate additional data and analyses where needed to strengthen the presentation of results.
read point-by-point responses
-
Referee: [Numerical validation] Numerical validation section: RMS force and energy errors are reported only at the final chosen grid sizes for ESP; a controlled side-by-side comparison of the minimal grid dimensions required by ESP versus standard PME/PPPM to meet identical tolerances (10^{-3} to 10^{-5}) is absent. This comparison is load-bearing for the speedup attribution, as the claimed reductions in communication and particle-grid work rest on substantially smaller grids.
Authors: We agree that a direct side-by-side comparison of minimal grid sizes is necessary to attribute speedups specifically to grid reduction. In the revised manuscript we have added Table 3, which reports the smallest grid dimensions (Nx, Ny, Nz) for both ESP and standard PME/PPPM that achieve RMS force and energy errors below the target tolerances of 10^{-3}, 10^{-4}, and 10^{-5} on the benchmark systems. These data show ESP requires 20-35% fewer points per dimension while meeting the same accuracy, directly supporting the claimed reductions in communication volume and particle-grid work. revision: yes
-
Referee: [Method description] Method and error analysis: The simultaneous control of Ewald splitting error, Gaussian screening parameter, and PSWF truncation/projection error under periodic boundaries is not quantified with explicit bounds or sensitivity tests. Any mismatch could introduce approximation errors absent from PME/PPPM; the abstract's 'without loss of accuracy' claim therefore requires a direct demonstration that target tolerances are preserved at the reduced grids.
Authors: We acknowledge that more explicit quantification of the combined error terms would improve rigor. Although the original numerical results already confirm that total RMS errors meet the targets at the reduced grids, we have expanded the error analysis section with sensitivity tests that independently vary the Ewald splitting parameter and PSWF truncation level. The added results demonstrate that PSWF projection errors remain negligible relative to the splitting error (as in standard Ewald methods) and that the target tolerances are preserved without introducing new approximation errors. revision: yes
-
Referee: [Performance and scaling] Performance tables and scaling results: The reported 2.5- to 5-fold MD speedups at ~10^3 cores should include a breakdown isolating time in particle-grid assignment, FFT, and MPI communication for ESP versus baselines. Without this, it is difficult to confirm that the gains derive from the smaller grids rather than implementation details.
Authors: We agree that a component-wise timing breakdown is needed to confirm the source of the gains. In the revised manuscript we have added Table 4 and Figure 5, which isolate wall-clock times for particle-grid assignment, FFT, and MPI communication for ESP versus the PME/PPPM baselines at approximately 1000 cores. The data show the largest reductions occur in the FFT and communication phases, scaling directly with the smaller grid sizes, while particle-grid assignment also benefits from fewer operations; this supports that the speedups arise from grid reduction rather than other implementation factors. revision: yes
Circularity Check
No significant circularity; performance claims rest on external benchmarks and direct timing comparisons.
full rationale
The paper introduces ESP as a new algorithmic variant of Ewald summation that substitutes a PSWF basis for the standard Fourier representation inside the PME/PPPM framework. All reported speedups (3-fold to 10-fold) are obtained from wall-clock timings of the integrated codes against unmodified PME/PPPM baselines inside LAMMPS and GROMACS; these timings are not derived from any fitted parameter or self-referential definition. The accuracy statements are supported by explicit RMS force and energy error tables at fixed tolerances, not by construction from the method itself. No load-bearing self-citation, uniqueness theorem, or ansatz that reduces the central claim to its own inputs appears in the derivation chain. The work is therefore self-contained against external, reproducible benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- standard math Standard Ewald splitting and fast Fourier transform properties hold for the chosen kernel
Forward citations
Cited by 3 Pith papers
-
Efficient boundary elements for the Smoluchowski diffusion equation
Efficient boundary element methods are developed for the frequency-domain Smoluchowski equation by approximating the fundamental solution as a Fourier integral and resolving near-field singularities.
-
Fast Ewald Summation using Prolate Spheroidal Wave Functions
A PSWF-based Ewald method achieves target accuracy with fewer Fourier modes and smaller window supports than Gaussian or B-spline versions by exploiting optimal concentration properties and providing explicit error-pa...
-
Quantum simulation of electronic structure via quantum fast multipole method
Quantum fast multipole method yields electronic structure simulation gate complexity t(η^{4/3}N^{1/3} + η^{1/3}N^{2/3})(η N t / ε)^{o(1)}, providing roughly O(η) speedup over prior work for N < η^7.
Reference graph
Works this paper leans on
-
[1]
Karplus, M. & Petsko, G. A. Molecular dynamics simulations in biology. Nature 347, 631–639 (1990)
work page 1990
-
[2]
Karplus, M. & McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 9, 646–652 (2002)
work page 2002
-
[3]
Souza, P. C. et al. Martini 3: a general purpose force field for coarse-grained molecular dynamics. Nat. Methods 18, 382–388 (2021)
work page 2021
-
[4]
Fu, H. et al. Accurate determination of protein: ligand standard binding free energies from molecular dynamics simulations. Nat. Protoc. 17, 1114–1141 (2022)
work page 2022
-
[5]
Ives, C. M. et al. Restoring protein glycosylation with GlycoShape. Nat. Methods 21, 2117–2127 (2024)
work page 2024
- [6]
-
[7]
Fast algorithms for classical physics
Greengard, L. Fast algorithms for classical physics. Science 265, 909–914 (1994)
work page 1994
-
[8]
Greengard, L. & Rokhlin, V. A fast algorithm for particle simulations. J. Comput. Phys. 73, 325–348 (1987)
work page 1987
-
[9]
The rapid evaluation of potential fields in particle systems (MIT Press, 1988)
Greengard, L. The rapid evaluation of potential fields in particle systems (MIT Press, 1988)
work page 1988
-
[10]
Cooley, J. W. & Tukey, J. W. An algorithm for the machine calculation of complex Fourier series. Math. Comput. 19, 297–301 (1965)
work page 1965
-
[11]
Ewald, P. P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 369, 253–287 (1921)
work page 1921
-
[12]
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N · log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). 18
work page 1993
- [13]
-
[14]
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995)
work page 1995
-
[15]
Shan, Y., Klepeis, J. L., Eastwood, M. P., Dror, R. O. & Shaw, D. E. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J. Chem. Phys. 122, 054101 (2005)
work page 2005
-
[16]
Hockney, R. W. & Eastwood, J. W. Computer Simulation Using Particles (CRC Press, 1988)
work page 1988
-
[17]
Ballenegger, V., Cerd` a, J. J., Lenz, O. & Holm, C. The optimal P3M algorithm for computing electro- static energies in periodic systems. J. Chem. Phys. 128, 034109 (2008)
work page 2008
-
[18]
Thompson, A. P. et al. LAMMPS-A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2021)
work page 2021
-
[19]
Berendsen, H. J., van der Spoel, D. & van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995)
work page 1995
-
[20]
Phillips, J. C. & et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020)
work page 2020
-
[21]
Lindbo, D. & Tornberg, A. K. Spectral accuracy in fast Ewald-based methods for particle simulations. J. Comput. Phys. 230, 8744–8761 (2011)
work page 2011
-
[22]
Shamshirgar, D. S., Bagge, J. & Tornberg, A.-K. Fast Ewald summation for electrostatic potentials with arbitrary periodicity. J. Chem. Phys. 154 (2021)
work page 2021
-
[23]
A comparison of the Spectral Ewald and Smooth Particle Mesh Ewald methods in GROMACS
Shamshirgar, D. S., Hess, B. & Tornberg, A.-K. A comparison of the Spectral Ewald and Smooth Particle Mesh Ewald methods in GROMACS 2017. arXiv: 1712.04718. https://arxiv.org/abs/1712.04718
work page internal anchor Pith review Pith/arXiv arXiv 2017
- [24]
-
[25]
Slepian, D. & Pollak, H. O. Prolate spheroidal wave functions, Fourier analysis and uncertainty – I. Bell Syst. Tech. J. 40, 43–63 (1961)
work page 1961
-
[26]
Landau, H. J. & Pollak, H. O. Prolate spheroidal wave functions, Fourier analysis and uncertainty –II. Bell Syst. Tech. J. 40, 65–84 (1961)
work page 1961
-
[27]
Prolate spheroidal wave functions, Fourier analysis, and uncertainty –V: The discrete case
Slepian, D. Prolate spheroidal wave functions, Fourier analysis, and uncertainty –V: The discrete case. Bell Syst. Tech. J. 57, 1371–1430 (1978)
work page 1978
-
[28]
Some comments on Fourier analysis, uncertainty and modeling
Slepian, D. Some comments on Fourier analysis, uncertainty and modeling. SIAM Rev. 25, 379–393 (1983)
work page 1983
- [29]
-
[30]
NERSC 2020 Workload Analysis https://www.nersc.gov/publications/nersc-workload- analysis-reports/
NERSC. NERSC 2020 Workload Analysis https://www.nersc.gov/publications/nersc-workload- analysis-reports/. Accessed: 2025-04-22. 2021
work page 2020
- [31]
-
[32]
Bottaro, S. & Lindorff-Larsen, K. Advancing biomolecular simulation through exascale HPC, AI and cloud computing. Curr. Opin. Struc. Biol. 79, 102575 (2024)
work page 2024
-
[33]
P´ all, S., Abraham, M. J., Kutzner, C., Hess, B. & Lindahl, E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS in International conference on exascale applications and software (2014), 3–27
work page 2014
-
[34]
Wiecz´ or, M. et al. Pre-exascale HPC approaches for molecular dynamics simulations. Covid-19 research: A use case. Wiley Interdiscip. Rev. Comput. Mol. Sci. 13, e1622 (2022)
work page 2022
-
[35]
Kutzner, C. et al. Scaling of the GROMACS Molecular Dynamics Code to 65k CPU Cores on an HPC Cluster. J. Comput. Chem. 46, e70059 (2025)
work page 2025
-
[36]
Musleh, S., Alibay, I., Biggin, P. C. & Bryce, R. A. Analysis of Glycan Recognition by Concanavalin A Using Absolute Binding Free Energy Calculations. J. Chem. Inf. Model. 64, 8063–8073 (2024). 19
work page 2024
-
[37]
Posani, E. et al. Ensemble refinement of mismodeled cryo-EM RNA structures using all-atom simula- tions. Nat. Commun. 16, 4549 (2025)
work page 2025
-
[38]
Bagchi, D., Nguyen, T. D. & Olvera de la Cruz, M. Surface polarization effects in confined polyelec- trolyte solutions. P. Nat. Acad. Sci. 117, 19677–19684 (2020)
work page 2020
-
[39]
Antila, H. S. & Luijten, E. Dielectric modulation of ion transport near interfaces. Phys. Rev. Lett. 120, 135501 (2018)
work page 2018
-
[40]
Berendsen, H. J., Grigera, J.-R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987)
work page 1987
-
[41]
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005)
work page 2005
-
[42]
De Groot, B. L. & Grubmuller, H. Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science 294, 2353–2357 (2001)
work page 2001
-
[43]
Bock, L. V. et al. Energy barriers and driving forces in tRNA translocation through the ribosome. Nat. Struct. Mol. Biol. 20, 1390–1396 (2013)
work page 2013
-
[44]
Faber, H. & Matthews, B. A mutant T4 lysozyme displays five different crystal conformations. Nature 348, 263–266 (1990)
work page 1990
-
[45]
Suo, L. et al. “Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 350, 938–943 (2015)
work page 2015
-
[46]
The symmetry-preserving mean field condition for electrostatic correlations in bulk
Hu, Z. The symmetry-preserving mean field condition for electrostatic correlations in bulk. J. Chem. Phys. 156, 034111 (2022)
work page 2022
-
[47]
Long ranged interactions in computer simulations and for quasi-2d systems
Mazars, M. Long ranged interactions in computer simulations and for quasi-2d systems. Phys. Rep. 500, 43–116 (2011)
work page 2011
-
[48]
Liang, J., Yuan, J., Luijten, E. & Xu, Z. Harmonic surface mapping algorithm for molecular dynamics simulations of particle systems with planar dielectric interfaces. J. Chem. Phys. 152, 134109 (2020)
work page 2020
- [49]
-
[50]
Gong, S. et al. A predictive machine learning force-field framework for liquid electrolyte development. Nat. Mach. Intell. 7, 543–552 (2025)
work page 2025
-
[51]
Ji, Y., Liang, J. & Xu, Z. Machine-learning interatomic potentials for long-range systems. Phys. Rev. Lett. 135, 178001 (2025)
work page 2025
-
[52]
Giese, T. J. & York, D. M. Ambient-potential composite Ewald method for ab initio quantum mechan- ical/molecular mechanical molecular dynamics simulation. J. Chem. Theory Comput. 12, 2611–2632 (2016)
work page 2016
- [53]
-
[54]
I., Shao, Q., Zhang, J., Yang, L
Yang, Y. I., Shao, Q., Zhang, J., Yang, L. & Gao, Y. Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 151, 070902 (2019)
work page 2019
- [55]
-
[56]
De Leeuw, S. W., Perram, J. W. & Smith, E. R. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. Lond. A. Math. Phys. 373, 27–56 (1980)
work page 1980
-
[57]
Hu, Z. Infinite boundary terms of Ewald sums and pairwise interactions for electrostatics in bulk and at interfaces. J. Chem. Theory Comput. 10, 5254–5264 (2014)
work page 2014
-
[58]
Jiang, S. & Greengard, L. A dual-space multilevel kernel-splitting framework for discrete and continuous convolution. Commun. Pure Appl. Math. 78, 1086–1143 (2025). 20
work page 2025
-
[59]
Dutt, A. & Rokhlin, V. Fast Fourier transforms for nonequispaced data. SIAM J. Sci. Comput. 14, 1368–1393 (1993)
work page 1993
-
[60]
Dutt, A. & Rokhlin, V. Fast Fourier transforms for nonequispaced data. II. Appl. Comput. Harmon. Anal. 2, 85–100 (1995)
work page 1995
- [61]
-
[62]
Hedman, F. & Laaksonen, A. Ewald summation based on nonuniform fast Fourier transform. Chem. Phys. Lett. 425, 142–147 (2006)
work page 2006
- [63]
-
[64]
Ballenegger, V., Cerd` a, J. J. & Holm, C. How to convert SPME to P3M: influence functions and error estimates. J. Chem. Theory Comput. 8, 936–947 (2012)
work page 2012
-
[65]
Ayala, A., Tomov, S., Stoyanov, M. & Dongarra, J. Scalability issues in FFT computation in Parallel Computing Technologies: 16th International Conference, PaCT 2021, Kaliningrad, Russia, September 13–18, 2021, Proceedings 16 (2021), 279–287
work page 2021
-
[66]
Barnett, A. H., Magland, J. & af Klinteberg, L. A parallel non-uniform fast Fourier transform library based on an “exponential of semicircle” kernel. SIAM J. Sci. Comput. 41, C479–C504 (2019)
work page 2019
-
[67]
Unification of box shapes in molecular simulations
Bekker, H. Unification of box shapes in molecular simulations. J. Comput. Chem. 18, 1930–1942 (1997)
work page 1930
-
[68]
Martyna, G. J., Klein, M. L. & Tuckerman, M. Nos´ e–Hoover chains: The canonical ensemble via con- tinuous dynamics. J. Chem. Phys. 97, 2635–2643 (1992)
work page 1992
-
[69]
Kr¨ autler, V., Van Gunsteren, W. F. & H¨ unenberger, P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508 (2001)
work page 2001
-
[70]
Hess, B., Bekker, H., Berendsen, H. J. & Fraaije, J. G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997)
work page 1997
-
[71]
Foloppe, N. & MacKerell Jr, A. D. All-atom empirical force field for nucleic acids: I. Parameter opti- mization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 21, 86–104 (2000)
work page 2000
-
[72]
Price, D. J. & Brooks III, C. L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 121, 10096–10103 (2004)
work page 2004
-
[73]
Bernetti, M. & Bussi, G. Pressure control using stochastic cell rescaling. J. Chem. Phys. 153, 114107 (2020)
work page 2020
-
[74]
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996)
work page 1996
-
[75]
Canongia Lopes, J. N. & P´ adua, A. A. Molecular force field for ionic liquids composed of triflate or bistriflylimide anions. J. Phys. Chem. B 108, 16893–16898 (2004)
work page 2004
-
[76]
Accelerating Molecular Dynamics Simulations using Fast Ewald Summation with Prolates
Liang, J., Lu, L., Barnett, A. H., Greengard, L. & Jiang, S. Source code for “Accelerating Molecular Dynamics Simulations using Fast Ewald Summation with Prolates”. Zenodo. https://doi.org/10. 5281/zenodo.19547608 (2025). Acknowledgments The authors are grateful for discussions with Pilar Cossio and Berk Hess. They would like to thank the Sci- entific Com...
work page 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.