Recognition: no theorem link
An Imbalanced Dataset with Multiple Feature Representations for Studying Quality Control of Next-Generation Sequencing
Pith reviewed 2026-05-13 16:58 UTC · model grok-4.3
The pith
A dataset of 37,491 NGS samples supplies two feature sets that let supervised models accurately recover expert quality labels.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that the supplied QC-34 features and the variable-length blocklist features both contain sufficient signal for supervised machine learning algorithms to predict the binary quality labels that were derived from automated tools and expert review, thereby confirming that the representations are relevant for automated quality control of next-generation sequencing data.
What carries the argument
Two parallel feature representations for the same samples—one a fixed vector of 34 quality-control metrics and the other a variable-length vector of read counts in ENCODE blocklist regions—paired with a binary quality label.
If this is right
- Direct side-by-side comparison of the 34-metric set versus the blocklist-derived set becomes possible for the same samples.
- Varying the number of blocklist features from eight to 1,183 lets researchers measure how feature granularity affects detection performance.
- The resource supports training and benchmarking of automated quality-control classifiers that must handle the observed 3.2 percent low-quality rate.
- Because all samples come from five genomic assays, performance differences across assay types can be quantified using identical feature pipelines.
Where Pith is reading between the lines
- Models trained on this data could be tested for transfer to sequencing platforms or species outside the current human and mouse collection.
- The extreme class imbalance suggests that future work should examine whether the reported accuracy holds when low-quality samples become even rarer in production runs.
- Combining the blocklist read-count features with base-level sequence models might reveal quality defects that neither representation captures alone.
Load-bearing premise
The binary quality labels assigned by existing automated tools plus domain experts constitute accurate ground truth that generalizes across experimental settings and assays.
What would settle it
Training the same supervised models on a new collection of NGS samples whose quality labels were assigned independently by multiple blinded experts and obtaining substantially lower prediction accuracy would falsify the claim.
Figures

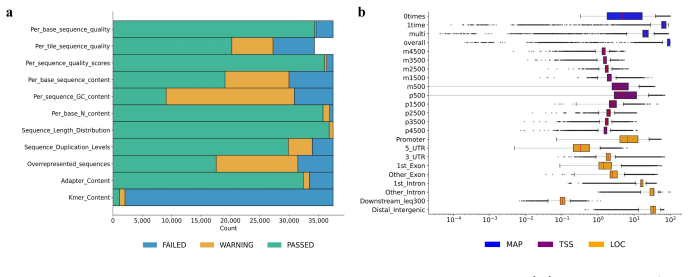
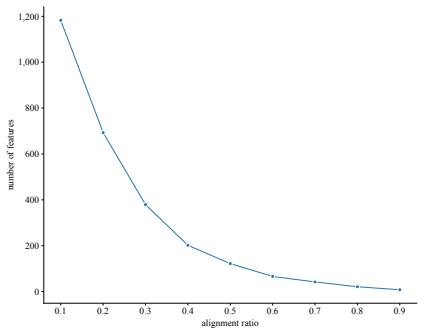

read the original abstract
Next-generation sequencing (NGS) is a key technique for studying the DNA and RNA of organisms. However, identifying quality problems in NGS data across different experimental settings remains challenging. To develop automated quality-control tools, researchers require datasets with features that capture the characteristics of quality problems. Existing NGS repositories, however, offer only a limited number of quality-related features. To address this gap, we propose a dataset derived from 37,491 NGS samples with two types of quality-related feature representations. The first type consists of 34 features derived from quality control tools (QC-34 features). The second type has a variable number of features ranging from eight to 1,183. These features were derived from read counts in problematic genomic regions identified by the ENCODE blocklist (BL features). All features describe the same human and mouse samples from five genomic assays, allowing direct comparison of feature representations. The proposed dataset includes a binary quality label, derived from automated quality control and domain experts. Among all samples, $3.2\%$ are of low quality. Supervised machine learning algorithms accurately predicted quality labels from the features, confirming the relevance of the provided feature representations. The proposed feature representations enable researchers to study how different feature types (QC-34 vs. BL features) and granularities (varying number of BL features) affect the detection of quality problems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a dataset of 37,491 NGS samples (human and mouse, five genomic assays) containing two feature representations—34 QC-derived features (QC-34) and variable-length BL features (8–1,183) extracted from ENCODE blocklist regions—together with binary quality labels (3.2 % low-quality) assigned from automated QC tools and domain experts. It reports that supervised ML models accurately predict these labels from the features, thereby confirming the utility of both representations for NGS quality-control research.
Significance. Release of a large, multi-assay dataset with two distinct feature families enables systematic comparison of QC-34 versus blocklist-derived representations at varying granularities. If the labels are shown to be independent of the QC-34 metrics, the resource would meaningfully support development and benchmarking of automated quality-control pipelines.
major comments (2)
- [Abstract] Abstract: the statement that supervised ML algorithms 'accurately predicted quality labels from the features, confirming the relevance' is unsupported by any reported performance metrics (accuracy, AUC, F1, etc.), train-test split details, cross-validation scheme, or imbalance-handling strategy. Without these numbers the confirmation claim cannot be evaluated.
- [Dataset construction and label derivation] Dataset construction and label derivation: QC-34 features are explicitly 'derived from quality control tools' while the binary label is also 'derived from automated quality control and domain experts.' If label assignment uses thresholds or rules on the same or linearly related QC metrics that populate QC-34, then high ML accuracy is expected by construction and does not independently validate feature relevance. The paper does not clarify the degree of overlap or provide an ablation that isolates the BL features from this risk.
minor comments (2)
- [Abstract] Abstract: the phrase 'a variable number of features ranging from eight to 1,183' should specify the exact mapping (e.g., which assays or samples receive which cardinalities) and the precise definition of the blocklist regions used.
- [Abstract] Abstract: the five genomic assays and the human/mouse split should be quantified (sample counts per assay and species) to allow readers to judge balance and generalizability.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and the recommendation for minor revision. We address each major comment below and have revised the manuscript to improve clarity and support for the claims.
read point-by-point responses
-
Referee: [Abstract] Abstract: the statement that supervised ML algorithms 'accurately predicted quality labels from the features, confirming the relevance' is unsupported by any reported performance metrics (accuracy, AUC, F1, etc.), train-test split details, cross-validation scheme, or imbalance-handling strategy. Without these numbers the confirmation claim cannot be evaluated.
Authors: We agree that the abstract would benefit from explicit quantitative support. We have revised the abstract to briefly report the key performance metrics from the ML experiments described in the main text, including AUC, F1-score for the low-quality class, the 80/20 train-test split, stratified 5-fold cross-validation, and the use of class weighting to address imbalance. These additions make the confirmation claim directly evaluable while preserving the abstract's brevity. revision: yes
-
Referee: [Dataset construction and label derivation] Dataset construction and label derivation: QC-34 features are explicitly 'derived from quality control tools' while the binary label is also 'derived from automated quality control and domain experts.' If label assignment uses thresholds or rules on the same or linearly related QC metrics that populate QC-34, then high ML accuracy is expected by construction and does not independently validate feature relevance. The paper does not clarify the degree of overlap or provide an ablation that isolates the BL features from this risk.
Authors: We thank the referee for highlighting this important potential circularity. The binary labels were assigned via a two-stage process: initial automated QC flags followed by independent expert review that incorporated additional criteria (e.g., visual inspection of coverage plots and experimental metadata) not reducible to the QC-34 values. To isolate the contribution of the BL features, we have added an ablation study to the revised manuscript in which models are trained using only the BL representation; these models retain substantial predictive performance. We have also expanded the methods section to explicitly describe the label derivation workflow and the limited direct overlap with QC-34 thresholds. revision: yes
Circularity Check
No circularity: empirical dataset release with standard ML validation
full rationale
The paper releases a dataset of NGS samples with QC-34 features (derived from quality control tools) and BL features, plus binary quality labels (from automated QC and domain experts). The central claim is the empirical observation that supervised ML algorithms accurately predict these labels from the features. No equations, parameter fitting, self-citations, or uniqueness theorems are invoked that would reduce the prediction result to the inputs by construction. The validation uses straightforward supervised learning on the released data and does not constitute a derivation chain. This is a normal, non-circular contribution for a dataset paper.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Quality labels produced by automated QC tools plus domain experts constitute reliable ground truth.
Reference graph
Works this paper leans on
-
[1]
Haley M Amemiya, Anshul Kundaje, and Alan P Boyle. The encode blacklist: identification of problematic regions of the genome.Scientific reports, 9(1):9354, 2019
work page 2019
-
[2]
Next-generation sequencing technology: current trends and advancements.Biology, 12(7):997, 2023
Heena Satam, Kandarp Joshi, Upasana Mangrolia, Sanober Waghoo, Gulnaz Zaidi, Shravani Rawool, Ritesh P Thakare, Shahid Banday, Alok K Mishra, Gautam Das, et al. Next-generation sequencing technology: current trends and advancements.Biology, 12(7):997, 2023
work page 2023
-
[3]
Timothé Ménard, Alaina Barros, and Christopher Ganter. Clinical quality considerations when using next-generation sequencing (ngs) in clinical drug development.Therapeutic Innovation & Regulatory Science, 55(5):1066– 1074, 2021
work page 2021
-
[4]
Margaret A Taub, Hector Corrada Bravo, and Rafael A Irizarry. Overcoming bias and systematic errors in next generation sequencing data.Genome medicine, 2:1–5, 2010
work page 2010
-
[5]
Maximilian Sprang, Jannik Möllmann, Miguel A Andrade-Navarro, and Jean-Fred Fontaine. Overlooked poor-quality patient samples in sequencing 19 data impair reproducibility of published clinically relevant datasets.Genome biology, 25(1):222, 2024
work page 2024
-
[6]
ENCODE Project Consortium. A user’s guide to the encyclopedia of dna elements (encode).PLoS Biology, 9(4):e1001046, 2011. doi: 10.1371/journal. pbio.1001046. URLhttps://doi.org/10.1371/journal.pbio.1001046
-
[7]
Sequencing Quality Control Consortium. A comprehensive assessment of rna-seq accuracy, reproducibility and information content by the sequencing quality control consortium.Nature biotechnology, 32(9):903–914, 2014
work page 2014
-
[8]
Mark D Wilkinson, Michel Dumontier, IJsbrand Jan Aalbersberg, Gabrielle Appleton, Myles Axton, Arie Baak, Niklas Blomberg, Jan-Willem Boiten, Luiz Bonino da Silva Santos, Philip E Bourne, et al. The fair guiding principles for scientific data management and stewardship.Scientific Data, 3(1):160018, 2016
work page 2016
-
[9]
Harrow, Rachel Drysdale, Andrew Smith, Susanna Repo, Jerry Lanfear, and Niklas Blomberg
Jennifer L. Harrow, Rachel Drysdale, Andrew Smith, Susanna Repo, Jerry Lanfear, and Niklas Blomberg. ELIXIR: providing a sustainable infrastruc- ture for life science data at european scale.Bioinform., 37(14):2506–2511, 2021
work page 2021
-
[10]
Andrade-Navarro, and Jean- Fred Fontaine
Steffen Albrecht, Maximilian Sprang, Miguel A. Andrade-Navarro, and Jean- Fred Fontaine. seqQscorer: automated quality control of next-generation sequencing data using machine learning.Genome Biology, 22(1):75, De- cember 2021. ISSN 1474-760X. doi: 10.1186/s13059-021-02294-2. URL https://genomebiology.biomedcentral.com/articles/10.1186/s13 059-021-02294-2
-
[11]
Jiajin Li, Brandon Jew, Lingyu Zhan, Sungoo Hwang, Giovanni Coppola, Nelson B. Freimer, and Jae Hoon Sul. Forestqc: Quality control on genetic variants from next-generation sequencing data using random forest.PLoS Comput. Biol., 15(12), 2019
work page 2019
-
[12]
Yunhai Luo, Benjamin C. Hitz, IdanGabdank, Jason A. Hilton, Meenakshi S. Kagda, Bonita Lam, Zachary A. Myers, Paul Sud, Jennifer Jou, Khine Lin, Ulugbek K. Baymuradov, Keenan Graham, Casey Litton, Stuart R. Miyasato, J. Seth Strattan, Otto Jolanki, Jin-Wook Lee, Forrest Tanaka, Philip Adenekan, Emma O’Neill, and J. Michael Cherry. New developments on the ...
work page 2020
-
[13]
An integrated encyclopedia of dna elements in the human genome.Nature, 489(7414):57, 2012
a) ENCODE Project Consortium et al. An integrated encyclopedia of dna elements in the human genome.Nature, 489(7414):57, 2012
work page 2012
-
[14]
Rongbin Zheng, Changxin Wan, Shenglin Mei, Qian Qin, Qiu Wu, Hanfei Sun, Chen-Hao Chen, Myles Brown, Xiaoyan Zhang, Clifford A. Meyer, and Xiaole Shirley Liu. Cistrome data browser: expanded datasets and new tools for gene regulatory analysis.Nucleic Acids Res., 47(Database-Issue): D729–D735, 2019. 20
work page 2019
-
[15]
How to overcome curse-of-dimensionality for out-of-distribution detection? In Michael J
Soumya Suvra Ghosal, Yiyou Sun, and Yixuan Li. How to overcome curse-of-dimensionality for out-of-distribution detection? In Michael J. Wooldridge, Jennifer G. Dy, and Sriraam Natarajan, editors,Thirty-Eighth AAAI Conference on Artificial Intelligence, AAAI 2024, Thirty-Sixth Con- ference on Innovative Applications of Artificial Intelligence, IAAI 2024, F...
-
[16]
Rna-seq: a revolutionary tool for transcriptomics.Nature reviews genetics, 10(1):57–63, 2009
Zhong Wang, Mark Gerstein, and Michael Snyder. Rna-seq: a revolutionary tool for transcriptomics.Nature reviews genetics, 10(1):57–63, 2009
work page 2009
-
[17]
Chip–seq: advantages and challenges of a maturing technology
Peter J Park. Chip–seq: advantages and challenges of a maturing technology. Nature reviews genetics, 10(10):669–680, 2009
work page 2009
-
[18]
Lingyun Song and Gregory E Crawford. Dnase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells.Cold Spring Harbor Protocols, 2010(2):pdb–prot5384, 2010
work page 2010
-
[19]
Robust transcriptome- wide discovery of rna-binding protein binding sites with enhanced clip (eclip)
Eric L Van Nostrand, Gabriel A Pratt, Alexander A Shishkin, Chelsea Gelboin-Burkhart, Mark Y Fang, Balaji Sundararaman, Steven M Blue, Thai B Nguyen, Christine Surka, Keri Elkins, et al. Robust transcriptome- wide discovery of rna-binding protein binding sites with enhanced clip (eclip). Nature methods, 13(6):508–514, 2016
work page 2016
-
[20]
Peter JA Cock, Christopher J Fields, Naohisa Goto, Michael L Heuer, and Peter M Rice. The sanger fastq file format for sequences with quality scores, and the solexa/illumina fastq variants.Nucleic acids research, 38(6): 1767–1771, 2010
work page 2010
-
[21]
Base- calling of automated sequencer traces using phred
Brent Ewing, LaDeana Hillier, Michael C Wendl, and Phil Green. Base- calling of automated sequencer traces using phred. i. accuracy assessment. Genome research, 8(3):175–185, 1998
work page 1998
-
[22]
Heng Li, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor T. Marth, Gonçalo R. Abecasis, and Richard Durbin. The sequence alignment/map format and samtools.Bioinform., 25(16):2078–2079, 2009
work page 2078
-
[23]
Fastqc: A quality control tool for high throughput sequence data
Simon Andrews. Fastqc: A quality control tool for high throughput sequence data. Available online athttp://www.bioinformatics.babraham.ac.uk/ projects/fastqc, 2010. Accessed: 2025-04-14
work page 2010
-
[24]
Fast gapped-read alignment with bowtie 2.Nature methods, 9(4):357–359, 2012
Ben Langmead and Steven L Salzberg. Fast gapped-read alignment with bowtie 2.Nature methods, 9(4):357–359, 2012
work page 2012
-
[25]
Maximilian Sprang, Matteo Krüger, Miguel A Andrade-Navarro, and Jean- Fred Fontaine. Statistical guidelines for quality control of next-generation 21 sequencing techniques.Life Science Alliance, 4(11):e202101113, November
-
[26]
ISSN 2575-1077. doi: 10.26508/lsa.202101113. URL https://www. life-science-alliance.org/lookup/doi/10.26508/lsa.202101113
-
[27]
Guangchuang Yu, Li-Gen Wang, and Qing-Yu He. Chipseeker: an r/bioconductor package for chip peak annotation, comparison and visu- alization.Bioinform., 31(14):2382–2383, 2015
work page 2015
-
[28]
Lihua Julie Zhu, Claude Gazin, Nathan D. Lawson, Hervé Pagès, Simon M. Lin, David S. Lapointe, and Michael R. Green. Chippeakanno: a biocon- ductor package to annotate chip-seq and chip-chip data.BMC Bioinform., 11:237, 2010
work page 2010
-
[29]
Diffbind: differential binding analysis of chip-seq peak data.R package version, 100(4.3):2–21, 2011
Rory Stark, Gordon Brown, et al. Diffbind: differential binding analysis of chip-seq peak data.R package version, 100(4.3):2–21, 2011
work page 2011
-
[30]
Model-based analysis of chip-seq (macs).Genome biology, 9:1–9, 2008
Yong Zhang, Tao Liu, Clifford A Meyer, Jérôme Eeckhoute, David S Johnson, Bradley E Bernstein, Chad Nusbaum, Richard M Myers, Myles Brown, Wei Li, et al. Model-based analysis of chip-seq (macs).Genome biology, 9:1–9, 2008
work page 2008
-
[31]
Steffen Albrecht, Clarissa Krämer, Philipp Röchner, Johannes U Mayer, Franz Rothlauf, Miguel A Andrade-Navarro, and Maximilian Sprang. Inte- grating the encode blocklist for machine learning quality control of chip-seq with seqqscorer.bioRxiv, 2025. doi: 10.1101/2025.05.12.653555. URLhttps: //www.biorxiv.org/content/early/2025/05/15/2025.05.12.653555
-
[32]
The encode (encyclopedia of dna elements) project.Science (New York, N.Y.), 306(5696):636–640, 2004
ENCODE Project Consortium. The encode (encyclopedia of dna elements) project.Science (New York, N.Y.), 306(5696):636–640, 2004. doi: 10.1126/ science.1105136. URLhttps://doi.org/10.1126/science.1105136
-
[33]
Kagda, Bonita Lam, Casey Litton, Corinn Small, Cricket A
Meenakshi S. Kagda, Bonita Lam, Casey Litton, Corinn Small, Cricket A. Sloan, Emma Spragins, Forrest Tanaka, Ian Whaling, Idan Gabdank, Ingrid Youngworth, J. Seth Strattan, Jason Hilton, Jennifer Jou, Jessica Au, Jin- Wook Lee, Kalina Andreeva, Keenan Graham, Khine Lin, Matt Simison, Otto Jolanki, Paul Sud, Pedro Assis, Philip Adenekan, Eric Douglas, Ming...
work page 2023
-
[34]
Jennifer Jou, Idan Gabdank, Yunhai Luo, Khine Lin, Paul Sud, Zachary Myers, Jason A. Hilton, Meenakshi S. Kagda, Bonita Lam, Emma O’Neill, Philip Adenekan, Keenan Graham, Ulugbek K. Baymuradov, Stuart R. Miyasato, J. Seth Strattan, Otto Jolanki, Jin-Wook Lee, Casey Lit- ton, Forrest Y. Tanaka, Benjamin C. Hitz, and J. Michael Cherry. The encode portal as ...
-
[35]
Cricket A. Sloan, Esther T. Chan, Jean M. Davidson, Venkat S. Malladi, J. Seth Strattan, Benjamin C. Hitz, Idan Gabdank, Aditi K. Narayanan, Marcus Ho, Brian T. Lee, Laurence D. Rowe, Timothy R. Dreszer, Greg Roe, Nikhil R. Podduturi, Forrest Tanaka, Eurie L. Hong, and J. Michael Cherry. ENCODE data at the ENCODE portal.Nucleic Acids Res., 44 (Database-Is...
work page 2016
-
[36]
Perspectives on ENCODE.Nature, 583(7818):693–698, July 2020
The ENCODE Project Consortium, Michael P Snyder, Thomas R Gingeras, Jill E Moore, Zhiping Weng, Mark B Gerstein, Bing Ren, Ross C Hardison, John A Stamatoyannopoulos, Brenton R Graveley, et al. Perspectives on ENCODE.Nature, 583(7818):693–698, July 2020. ISSN 0028-0836, 1476-
work page 2020
-
[37]
URL https://www.nature.com/a rticles/s41586-020-2449-8
doi: 10.1038/s41586-020-2449-8. URL https://www.nature.com/a rticles/s41586-020-2449-8
-
[38]
Aaron R. Quinlan and Ira M. Hall. Bedtools: a flexible suite of utilities for comparing genomic features.Bioinform., 26(6):841–842, 2010
work page 2010
-
[39]
The ucsc genome browser database: 2025 update.Nucleic Acids Research, 53(D1):D1243–D1249, 2025
Gerardo Perez, Galt P Barber, Anna Benet-Pages, Jonathan Casper, Hiram Clawson, Mark Diekhans, Clay Fischer, Jairo Navarro Gonzalez, Angie S Hinrichs, Christopher M Lee, et al. The ucsc genome browser database: 2025 update.Nucleic Acids Research, 53(D1):D1243–D1249, 2025
work page 2025
-
[40]
Tao Liu, Jorge A. Ortiz, Len Taing, Clifford A. Meyer, Bernett Lee, Yong Zhang, Hyunjin Shin, Swee S. Wong, Jian Ma, Ying Lei, Utz J. Pape, Michael Poidinger, Yiwen Chen, Kevin Yeung, Myles Brown, Yaron Turpaz, and X. Shirley Liu. Cistrome: an integrative platform for transcriptional regulation studies.Genome Biology, 12(8):R83, August 2011. ISSN 1474- 76...
-
[41]
McKnight and Julius Najab.Mann-Whitney U Test
Patrick E. McKnight and Julius Najab.Mann-Whitney U Test. John Wiley & Sons, Ltd, 2010. ISBN 9780470479216. doi: https://doi.org/10.1002/97 80470479216.corpsy0524. URL https://onlinelibrary.wiley.com/doi/ abs/10.1002/9780470479216.corpsy0524
work page doi:10.1002/97 2010
-
[42]
Sture Holm. A simple sequentially rejective multiple test procedure.Scan- dinavian Journal of Statistics, 6(2):65–70, 1979. ISSN 03036898, 14679469. URLhttp://www.jstor.org/stable/4615733
-
[43]
Andrew P. Bradley. The use of the area under the ROC curve in the evaluation of machine learning algorithms.Pattern Recognit., 30(7):1145– 1159, 1997
work page 1997
-
[44]
Friedman.The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd Edition
Trevor Hastie, Robert Tibshirani, and Jerome H. Friedman.The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd Edition. Springer Series in Statistics. Springer, 2009. 23
work page 2009
- [45]
-
[46]
Jerome H Friedman. Greedy function approximation: a gradient boosting machine.Annals of statistics, pages 1189–1232, 2001
work page 2001
-
[47]
MIT Press, 2016.http://www.deeplearningbook.org
Ian Goodfellow, Yoshua Bengio, and Aaron Courville.Deep Learning. MIT Press, 2016.http://www.deeplearningbook.org
work page 2016
-
[48]
Scikit-learn: Machine learning in python.J
Fabian Pedregosa, Gaël Varoquaux, Alexandre Gramfort, Vincent Michel, Bertrand Thirion, Olivier Grisel, Mathieu Blondel, Peter Prettenhofer, Ron Weiss, Vincent Dubourg, Jake VanderPlas, Alexandre Passos, David Cournapeau, Matthieu Brucher, Matthieu Perrot, and Edouard Duchesnay. Scikit-learn: Machine learning in python.J. Mach. Learn. Res., 12:2825– 2830, 2011
work page 2011
-
[49]
Diederik P. Kingma and Jimmy Ba. Adam: A method for stochastic optimization. InICLR (Poster), 2015. 24
work page 2015
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.