Recognition: unknown
Making Room for AI: Multi-GPU Molecular Dynamics with Deep Potentials in GROMACS
Pith reviewed 2026-05-10 17:02 UTC · model grok-4.3
The pith
GROMACS now supports production-scale molecular dynamics with deep neural network potentials on multi-GPU systems.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors add a DeePMD backend to the GROMACS NNPot interface and introduce a domain decomposition layer that decouples inference from the main simulation loop. Two MPI collectives handle coordinate broadcast and force redistribution each step, allowing concurrent GPU-accelerated inference on all ranks. They train a 1.6-million-parameter DPA-1 model on solvated protein fragments, validate it on small systems, and benchmark scaling on up to 32 A100 and MI250x GPUs, concluding that production MD with near ab initio fidelity is feasible at scale in GROMACS.
What carries the argument
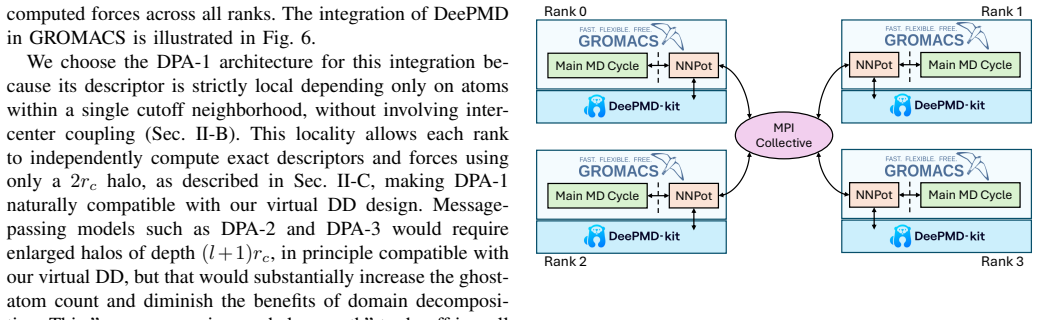
DeePMD backend for the GROMACS NNPot interface with a decoupled domain decomposition layer that uses two MPI collectives per step to exchange coordinates and forces while running inference concurrently on all processes.
If this is right
- Production molecular dynamics of solvated proteins with near ab initio accuracy becomes practical on existing GROMACS installations using 16 to 32 GPUs.
- Strong scaling reaches 66 percent efficiency at 16 devices and weak scaling reaches 80 percent at 16 devices for 15,000-atom systems.
- More than 90 percent of wall time is spent in DeePMD inference while MPI collectives contribute less than 10 percent.
- Irreducible ghost-atom costs set by the cutoff radius and load imbalance across ranks are the main scaling limits.
Where Pith is reading between the lines
- The same decoupled inference layer could be reused to plug in other neural-network potentials without re-engineering the GROMACS core.
- For systems much larger than 15,000 atoms the ghost-atom overhead may require shorter cutoffs or hybrid classical-AI potential schemes to maintain efficiency.
- Low MPI overhead suggests the approach would transfer to other distributed MD codes facing similar inference integration needs.
Load-bearing premise
The trained DPA-1 model provides forces accurate enough for the target protein systems and the combined inference plus MPI overhead stays low enough for long production simulations.
What would settle it
A direct comparison of long protein trajectories generated by the integrated GROMACS-DeePMD code against reference ab initio or experimental data that shows systematic deviations in structure or dynamics beyond acceptable error thresholds for the application.
Figures







read the original abstract
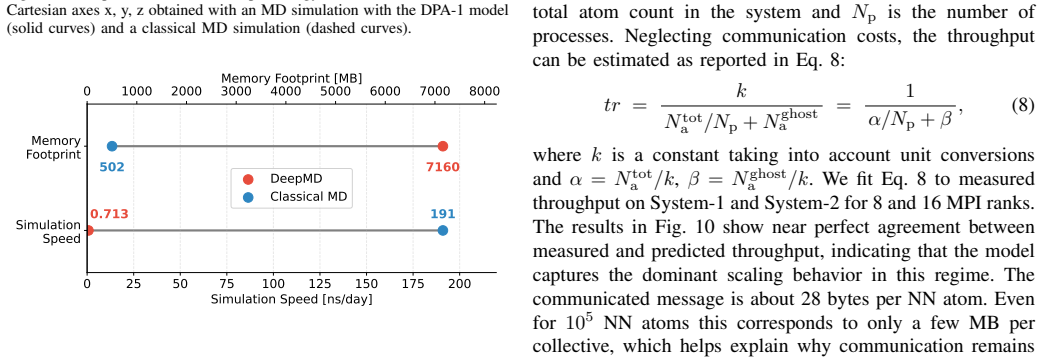
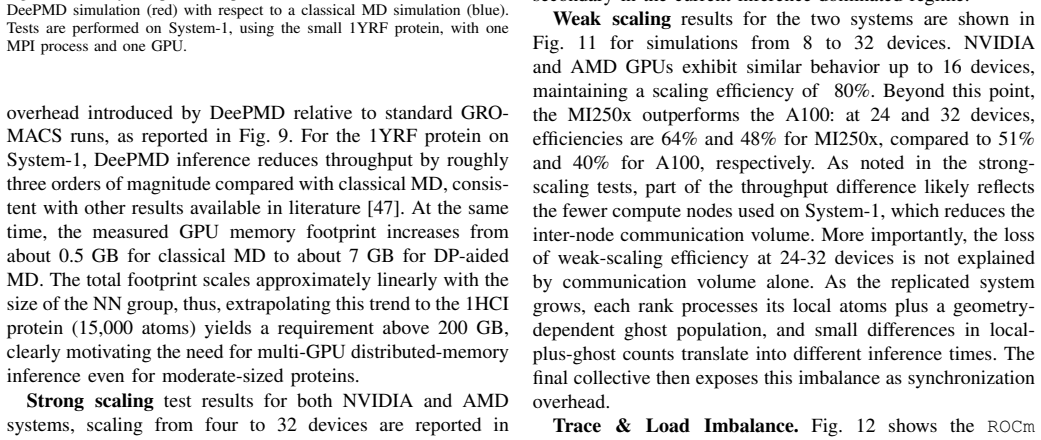
GROMACS is a de-facto standard for classical Molecular Dynamics (MD). The rise of AI-driven interatomic potentials that pursue near-quantum accuracy at MD throughput now poses a significant challenge: embedding neural-network inference into multi-GPU simulations retaining high-performance. In this work, we integrate the MLIP framework DeePMD-kit into GROMACS, enabling domain-decomposed, GPU-accelerated inference across multi-node systems. We extend the GROMACS NNPot interface with a DeePMD backend, and we introduce a domain decomposition layer decoupled from the main simulation. The inference is executed concurrently on all processes, with two MPI collectives used each step to broadcast coordinates and to aggregate and redistribute forces. We train an in-house DPA-1 model (1.6 M parameters) on a dataset of solvated protein fragments. We validate the implementation on a small protein system, then we benchmark the GROMACS-DeePMD integration with a 15,668 atom protein on NVIDIA A100 and AMD MI250x GPUs up to 32 devices. Strong-scaling efficiency reaches 66% at 16 devices and 40% at 32; weak-scaling efficiency is 80% to 16 devices and reaches 48% (MI250x) and 40% (A100) at 32 devices. Profiling with the ROCm System profiler shows that >90% of the wall time is spent in DeePMD inference, while MPI collectives contribute <10%, primarily since they act as a global synchronization point. The principal bottlenecks are the irreducible ghost-atom cost set by the cutoff radius, confirmed by a simple throughput model, and load imbalance across ranks. These results demonstrate that production MD with near ab initio fidelity is feasible at scale in GROMACS.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper describes the integration of DeePMD-kit into GROMACS via an extended NNPot interface and a decoupled domain-decomposition layer, using two MPI collectives per step for coordinate broadcast and force aggregation/redistribution. An in-house 1.6 M parameter DPA-1 model is trained on solvated protein fragments; the implementation is validated on a small protein and then benchmarked on a 15,668-atom protein system using up to 32 NVIDIA A100 and AMD MI250x GPUs. Reported results include strong-scaling efficiencies of 66 % at 16 devices and 40 % at 32 devices, weak-scaling efficiencies of 40–48 % at 32 devices, profiling showing >90 % of wall time in DeePMD inference, and a simple throughput model identifying ghost-atom cutoff costs and load imbalance as principal bottlenecks. The work concludes that production MD with near ab initio fidelity is feasible at scale in GROMACS.
Significance. If the accuracy of the DPA-1 model is demonstrated, the integration would enable large-scale, high-fidelity molecular dynamics in a widely used production MD package, lowering the barrier for near-quantum-accuracy simulations of biomolecular systems. The concrete scaling numbers, ROCm profiling data, and simple throughput model provide practical, reproducible guidance for similar NN-potential integrations; these elements ground the performance claims and constitute a clear strength of the manuscript.
major comments (1)
- [Abstract and validation description] Abstract and validation description: the central claim that the integration 'demonstrates that production MD with near ab initio fidelity is feasible at scale' is not supported by any quantitative accuracy metrics for the trained DPA-1 model. No energy or force RMSE values, test-set statistics, or direct comparisons to DFT references are reported for the solvated protein fragments, the small validation protein, or the 15,668-atom benchmark system. This omission is load-bearing because the 'near ab initio fidelity' half of the feasibility claim rests entirely on unshown model accuracy rather than on the reported throughput results.
minor comments (2)
- [Profiling and throughput model] The simple throughput model is referenced but not shown or derived in sufficient detail to allow independent verification of the ghost-atom cost analysis.
- [Benchmarking results] Scaling efficiencies are reported as single-point values without error bars, number of repeated runs, or discussion of run-to-run variability.
Simulated Author's Rebuttal
We thank the referee for the thorough review and for highlighting this important issue with the support for our central claim. We respond point-by-point below.
read point-by-point responses
-
Referee: [Abstract and validation description] Abstract and validation description: the central claim that the integration 'demonstrates that production MD with near ab initio fidelity is feasible at scale' is not supported by any quantitative accuracy metrics for the trained DPA-1 model. No energy or force RMSE values, test-set statistics, or direct comparisons to DFT references are reported for the solvated protein fragments, the small validation protein, or the 15,668-atom benchmark system. This omission is load-bearing because the 'near ab initio fidelity' half of the feasibility claim rests entirely on unshown model accuracy rather than on the reported throughput results.
Authors: We agree with the referee that the manuscript contains no quantitative accuracy metrics (energy/force RMSE, test-set statistics, or DFT comparisons) for the in-house DPA-1 model on any of the systems mentioned. The paper's core contribution is the software integration (extended NNPot interface, decoupled domain-decomposition layer, and two-MPI-collective communication pattern) together with the multi-GPU scaling results and throughput model. Model training details are provided only to describe the benchmark workload; the 'near ab initio fidelity' phrasing in the abstract and conclusions is therefore not backed by data shown here. We will revise the abstract, introduction, and conclusions to qualify the claim, stating that the integration enables production-scale MD at the fidelity of the trained deep potential (whose accuracy rests on its DFT training data). We will also add a brief clarifying sentence in the methods section. These changes will be incorporated in the revised manuscript. revision: yes
Circularity Check
No circularity: implementation paper with direct hardware benchmarks and no derivation chain
full rationale
The manuscript is an engineering report on integrating DeePMD-kit into GROMACS via an extended NNPot interface, introducing a decoupled domain-decomposition layer, and executing concurrent inference with two MPI collectives. It reports training a 1.6 M parameter DPA-1 model on solvated protein fragments, validation on a small system, and measured scaling efficiencies (strong scaling 66 % at 16 devices, 40 % at 32; weak scaling 40–48 % at 32 devices) plus profiling (>90 % time in inference) on A100 and MI250x hardware. No equations, fitted parameters renamed as predictions, self-citations forming load-bearing uniqueness arguments, or ansatzes smuggled via prior work appear. All performance numbers are direct wall-time measurements from GPU runs, not reductions to the paper’s own inputs. The assertion of “near ab initio fidelity” rests on the external deep-potential framework rather than any internal derivation that collapses by construction.
Axiom & Free-Parameter Ledger
free parameters (1)
- DPA-1 model parameters
axioms (1)
- domain assumption Standard molecular dynamics assumptions remain valid when replacing classical potentials with the DeePMD neural network potential.
Reference graph
Works this paper leans on
-
[1]
GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit,
S. Pronk, S. P ´all, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess, and E. Lindahl, “GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit,” Bioinformatics, vol. 29, no. 7, pp. 845–854, 02 2013. [Online]. Available: https://doi.org/10.1093/bioinfor...
-
[2]
GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers,
M. J. Abraham, T. Murtola, R. Schulz, S. P ´all, J. C. Smith, B. Hess, and E. Lindahl, “GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers,”Soft- wareX, vol. 1-2, pp. 19–25, 2015
2015
-
[3]
Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS,
S. P ´all, A. Zhmurov, P. Bauer, M. Abraham, M. Lundborg, A. Gray, B. Hess, and E. Lindahl, “Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS,”The Journal of Chemical Physics, vol. 153, no. 13, p. 134110, 10 2020. [Online]. Available: https://doi.org/10.1063/5.0018516
-
[4]
W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman, “A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules,”Journal of the American Chemical Society, vol. 117, no. 19, pp. 5179–5197, 1995. [Online]. Available: https://do...
-
[5]
All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins,
A. D. M. Jr., D. Bashford, M. Bellott, R. L. D. Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wi ´orkiewicz-Kuczera, D. Yin, and ...
-
[6]
Deep dive into machine learning density functional theory for materials science and chemistry,
L. Fiedler, K. Shah, M. Bussmann, and A. Cangi, “Deep dive into machine learning density functional theory for materials science and chemistry,”Phys. Rev. Mater., vol. 6, p. 040301, Apr 2022
2022
-
[7]
A practical guide to machine learning interatomic potentials – Status and future,
R. Jacobs, D. Morgan, S. Attarian, J. Meng, C. Shen, Z. Wu, C. Y . Xie, J. H. Yang, N. Artrith, B. Blaiszik, G. Ceder, K. Choudhary, G. Csanyi, E. D. Cubuk, B. Deng, R. Drautz, X. Fu, J. Godwin, V . Honavar, O. Isayev, A. Johansson, B. Kozinsky, S. Martiniani, S. P. Ong, I. Poltavsky, K. Schmidt, S. Takamoto, A. P. Thompson, J. Westermayr, and B. M. Wood,...
2025
-
[8]
DeePMD-kit v3: A Multiple-Backend Framework for Machine Learning Potentials,
J. Zeng, D. Zhang, A. Peng, X. Zhang, S. He, Y . Wang, X. Liu, H. Bi, Y . Li, C. Cai, C. Zhang, Y . Du, J.-X. Zhu, P. Mo, Z. Huang, Q. Zeng, S. Shi, X. Qin, Z. Yu, C. Luo, Y . Ding, Y .-P. Liu, R. Shi, Z. Wang, S. L. Bore, J. Chang, Z. Deng, Z. Ding, S. Han, W. Jiang, G. Ke, Z. Liu, D. Lu, K. Muraoka, H. Oliaei, A. K. Singh, H. Que, W. Xu, Z. Xu, Y .-B. Z...
-
[9]
M. P. Allen and D. J. Tildesley,Computer Simulation of Liquids. Oxford University Press, 06 2017. [Online]. Available: https: //doi.org/10.1093/oso/9780198803195.001.0001
-
[10]
Zhou and B
K. Zhou and B. Liu,Molecular Dynamics Simulation: Fundamentals and Applications, 1st ed. Academic Press, 2022
2022
-
[11]
Ab initio molecular dy- namics: Concepts, recent developments, and future trends,
R. Iftimie, P. Minary, and M. E. Tuckerman, “Ab initio molecular dy- namics: Concepts, recent developments, and future trends,”Proceedings of the National Academy of Sciences, vol. 102, no. 19, pp. 6654–6659, 2005
2005
-
[12]
Towards electronic structure-based ab-initio molecular dynamics simulations with hundreds of millions of atoms,
R. Schade, T. Kenter, H. Elgabarty, M. Lass, O. Sch ¨utt, A. Lazzaro, H. Pabst, S. Mohr, J. Hutter, T. D. K ¨uhne, and C. Plessl, “Towards electronic structure-based ab-initio molecular dynamics simulations with hundreds of millions of atoms,”Parallel Computing, vol. 111, p. 102920, 2022
2022
-
[13]
Machine Learn- ing for Molecular Simulation,
F. No ´e, A. Tkatchenko, K.-R. M¨uller, and C. Clementi, “Machine Learn- ing for Molecular Simulation,”Annual Review of Physical Chemistry, vol. 71, no. V olume 71, 2020, pp. 361–390, 2020
2020
-
[14]
DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics,
H. Wang, L. Zhang, J. Han, and W. E, “DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics,”Computer Physics Communications, vol. 228, pp. 178–184, 2018. [Online]. Available: https://www.sciencedirect.com/ science/article/pii/S0010465518300882
2018
-
[15]
End-to- end symmetry preserving inter-atomic potential energy model for finite and extended systems,
L. Zhang, J. Han, H. Wang, W. A. Saidi, R. Car, and E. Weinan, “End-to- end symmetry preserving inter-atomic potential energy model for finite and extended systems,” p. 4441–4451, 2018
2018
-
[16]
Pretraining of attention-based deep learning potential model for molecular simulation,
D. Zhang, H. Bi, F.-Z. Dai, W. Jiang, X. Liu, L. Zhang, and H. Wang, “Pretraining of attention-based deep learning potential model for molecular simulation,”npj Computational Materials, vol. 10, no. 1, p. 94, 2024. [Online]. Available: https://doi.org/10.1038/ s41524-024-01278-7
2024
-
[17]
DPA-2: a large atomic model as a multi-task learner,
D. Zhang, X. Liu, X. Zhang, C. Zhang, C. Cai, H. Bi, Y . Du, X. Qin, A. Peng, J. Huang, B. Li, Y . Shan, J. Zeng, Y . Zhang, S. Liu, Y . Li, J. Chang, X. Wang, S. Zhou, J. Liu, X. Luo, Z. Wang, W. Jiang, J. Wu, Y . Yang, J. Yang, M. Yang, F.-Q. Gong, L. Zhang, M. Shi, F.-Z. Dai, D. M. York, S. Liu, T. Zhu, Z. Zhong, J. Lv, J. Cheng, W. Jia, M. Chen, G. Ke...
-
[18]
Zhang et al., A Graph Neural Network for the Era of Large Atomistic Models
D. Zhang, A. Peng, C. Cai, W. Li, Y . Zhou, J. Zeng, M. Guo, C. Zhang, B. Li, H. Jiang, T. Zhu, W. Jia, L. Zhang, and H. Wang, “A Graph Neural Network for the Era of Large Atomistic Models,” 2025. [Online]. Available: https://arxiv.org/abs/2506.01686
-
[19]
A flexible algorithm for calculating pair interac- tions on SIMD architectures,
S. P ´all and B. Hess, “A flexible algorithm for calculating pair interac- tions on SIMD architectures,”Computer Physics Communications, vol. 184, no. 12, pp. 2641–2650, 2013
2013
-
[20]
General Hartree-Fock program.Computer Physics Com- munications43, 355–365 (1987)
S. Liem, D. Brown, and J. Clarke, “Molecular dynamics simulations on distributed memory machines,”Computer Physics Communications, vol. 67, no. 2, pp. 261–267, 1991. [Online]. Available: https: //www.sciencedirect.com/science/article/pii/001046559190021C
-
[21]
The midpoint method for parallelization of particle simulations,
K. J. Bowers, R. O. Dror, and D. E. Shaw, “The midpoint method for parallelization of particle simulations,”The Journal of Chemical Physics, vol. 124, no. 18, p. 184109, 05 2006. [Online]. Available: https://doi.org/10.1063/1.2191489
-
[22]
GROMACS Reference Manual,
GROMACS Development Team, “GROMACS Reference Manual,” https://manual.gromacs.org/current/reference-manual/index.html, 2025
2025
-
[23]
Redesign- ing GROMACS Halo Exchange: Improving Strong Scaling with GPU- initiated NVSHMEM,
M. Doijade, A. Alekseenko, A. Brown, A. Gray, and S. P ´all, “Redesign- ing GROMACS Halo Exchange: Improving Strong Scaling with GPU- initiated NVSHMEM,”arXiv preprint arXiv:2509.21527, 2025
-
[24]
Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials,
A. Thompson, L. Swiler, C. Trott, S. Foiles, and G. Tucker, “Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials,”Journal of Computational Physics, vol. 285, pp. 316–330, 2015. [Online]. Available: https: //www.sciencedirect.com/science/article/pii/S0021999114008353
2015
-
[25]
SIMPLE-NN: An efficient package for training and executing neural-network interatomic potentials,
K. Lee, D. Yoo, W. Jeong, and S. Han, “SIMPLE-NN: An efficient package for training and executing neural-network interatomic potentials,”Computer Physics Communications, vol. 242, pp. 95– 103, 2019. [Online]. Available: https://www.sciencedirect.com/science/ article/pii/S0010465519301298
2019
-
[26]
An implementation of artificial neural-network potentials for atomistic materials simulations: Performance for TiO2,
N. Artrith and A. Urban, “An implementation of artificial neural-network potentials for atomistic materials simulations: Performance for TiO2,” Computational Materials Science, vol. 114, pp. 135–150, 2016
2016
-
[27]
TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials,
X. Gao, F. Ramezanghorbani, O. Isayev, J. S. Smith, and A. E. Roitberg, “TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials,”Journal of Chemical Information and Modeling, vol. 60, no. 7, pp. 3408–3415, Jul 2020
2020
-
[28]
TorchMD: A deep learning framework for molecular simulations,
S. Doerr, M. Majewski, A. P ´erez, A. Kramer, C. Clementi, F. Noe, T. Giorgino, and G. De Fabritiis, “TorchMD: A deep learning framework for molecular simulations,”Journal of chemical theory and computation, vol. 17, no. 4, pp. 2355–2363, 2021
2021
-
[29]
TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations,
R. P. Pel ´aez, G. Simeon, R. Galvelis, A. Mirarchi, P. Eastman, S. Doerr, P. Th ¨olke, T. E. Markland, and G. D. Fabritiis, “TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations,”Journal of Chemical Theory and Computation, vol. 20, no. 10, pp. 4076–4087,
-
[30]
Available: https://doi.org/10.1021/acs.jctc.4c00253
[Online]. Available: https://doi.org/10.1021/acs.jctc.4c00253
-
[31]
Andreas Bender, Nadine Schneider, Marwin Segler, W
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, “E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials,” Nature Communications, vol. 13, no. 1, p. 2453, May 2022. [Online]. Available: https://doi.org/10.1038/s41467-022-29939-5
-
[32]
Learning local equivariant representations for large-scale atomistic dynamics,
A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth, and B. Kozinsky, “Learning local equivariant representations for large-scale atomistic dynamics,”Nature Communications, vol. 14, no. 1, p. 579, Feb 2023. [Online]. Available: https://doi.org/10.1038/s41467-023-36329-y
-
[33]
Materials Learning Algorithms (MALA): Scalable machine learning for electronic structure calculations in large-scale atomistic simulations,
A. Cangi, L. Fiedler, B. Brzoza, K. Shah, T. J. Callow, D. Kotik, S. Schmerler, M. C. Barry, J. M. Goff, A. Rohskopf, D. J. V ogel, N. Modine, A. P. Thompson, and S. Rajamanickam, “Materials Learning Algorithms (MALA): Scalable machine learning for electronic structure calculations in large-scale atomistic simulations,”Computer Physics Communications, vol...
2025
-
[34]
CHARMM at 45: Enhancements in Accessibility, Functionality, and Speed,
W. Hwang, S. L. Austin, A. Blondel, E. D. Boittier, S. Boresch, M. Buck, J. Buckner, A. Caflisch, H. Chang, X. Cheng, Y . K. Choi, J. Chu, M. F. Crowley, Q. Cui, A. Damjanovic, Y . Deng, M. Devereux, X. Ding, M. F. Feig, J. Gao, D. R. Glowacki, J. E. G. II, M. B. Hamaneh, E. D. Harder, R. L. Hayes, J. Huang, Y . Huang, P. S. Hudson, W. Im, S. M. Islam, W....
-
[35]
Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/Molecular Mechanical Simulations of Chemical Reactions in Solution,
J. Zeng, T. J. Giese, S ¸. Ekesan, and D. M. York, “Development of Range-Corrected Deep Learning Potentials for Fast, Accurate Quantum Mechanical/Molecular Mechanical Simulations of Chemical Reactions in Solution,”Journal of Chemical Theory and Computation, vol. 17, no. 11, pp. 6993–7009, Nov 2021
2021
-
[36]
D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros, V . W. D. Cruzeiro, N. Forouzesh, T. J. Giese, A. W. G ¨otz, H. Gohlke, S. Izadi, K. Kasavajhala, M. C. Kaymak, E. King, T. Kurtzman, T. Lee, P. Li, J. Liu, T. Luchko, R. Luo, M. Manathunga, M. R. Machado, H. M. Nguyen, K. A. O’Hearn, A. V . Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnam...
-
[37]
Implementation and Validation of an OpenMM Plugin for the Deep Potential Representation of Potential Energy,
Y . Ding and J. Huang, “Implementation and Validation of an OpenMM Plugin for the Deep Potential Representation of Potential Energy,” International Journal of Molecular Sciences, vol. 25, no. 3, 2024
2024
-
[38]
LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales,
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, “LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales,”Computer Physics Commu...
2022
-
[39]
AENET–LAMMPS and AENET–TINKER: interfaces for accurate and efficient molecular dynamics simulations with machine learning poten- tials,
M. S. Chen, T. Morawietz, H. Mori, T. E. Markland, and N. Artrith, “AENET–LAMMPS and AENET–TINKER: interfaces for accurate and efficient molecular dynamics simulations with machine learning poten- tials,”The Journal of Chemical Physics, vol. 155, no. 7, 2021
2021
-
[40]
MLMOD: Machine Learning Methods for Data-Driven Modeling in LAMMPS,
P. Atzberger, “MLMOD: Machine Learning Methods for Data-Driven Modeling in LAMMPS,”Journal of Open Source Software, vol. 8, p. 5620, 09 2023
2023
-
[41]
P. Fuchs, W. Chen, S. Thaler, and J. Zavadlav, “chemtrain-deploy: A parallel and scalable framework for machine learning potentials in million-atom MD simulations,”arXiv preprint arXiv:2506.04055, 2025
-
[42]
K. Nguyen-Cong, J. T. Willman, S. G. Moore, A. B. Belonoshko, R. Gayatri, E. Weinberg, M. A. Wood, A. P. Thompson, and I. I. Oleynik, “Billion atom molecular dynamics simulations of carbon at extreme conditions and experimental time and length scales,” in Proceedings of the International Conference for High Performance Computing, Networking, Storage and A...
-
[43]
Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning,
W. Jia, H. Wang, M. Chen, D. Lu, L. Lin, R. Car, W. E, and L. Zhang, “Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning,” inProceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, ser. SC ’20. IEEE Press, 2020
2020
-
[44]
Extending the limit of molecular dynamics with ab initio accuracy to 10 billion atoms,
Z. Guo, D. Lu, Y . Yan, S. Hu, R. Liu, G. Tan, N. Sun, W. Jiang, L. Liu, Y . Chen, L. Zhang, M. Chen, H. Wang, and W. Jia, “Extending the limit of molecular dynamics with ab initio accuracy to 10 billion atoms,” inProceedings of the 27th ACM SIGPLAN Symposium on Principles and Practice of Parallel Programming, ser. PPoPP ’22. New York, NY , USA: Associati...
-
[45]
J. Li, B. Li, Z. Guo, M. Li, E. Li, L. Liu, G. Yuan, Z. Wang, G. Tan, and W. Jia, “Scaling Molecular Dynamics with ab initio Accuracy to 149 Nanoseconds per Day,” inProceedings of the International Conference for High Performance Computing, Networking, Storage, and Analysis, ser. SC ’24. IEEE Press, 2024. [Online]. Available: https://doi.org/10.1109/SC414...
-
[46]
MACE: higher order equivariant message passing neural networks for fast and accurate force fields,
I. Batatia, D. P. Kov ´acs, G. N. C. Simm, C. Ortner, and G. Cs ´anyi, “MACE: higher order equivariant message passing neural networks for fast and accurate force fields,” inProceedings of the 36th International Conference on Neural Information Processing Systems, ser. NIPS ’22. Red Hook, NY , USA: Curran Associates Inc., 2022
2022
-
[47]
PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges,
O. T. Unke and M. Meuwly, “PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges,”Journal of Chemical Theory and Computation, vol. 15, no. 6, pp. 3678–3693, Jun
-
[48]
Available: https://doi.org/10.1021/acs.jctc.9b00181
[Online]. Available: https://doi.org/10.1021/acs.jctc.9b00181
-
[49]
Enabling AI Deep Potentials for Ab Initio-quality Molecular Dynamics Simulations in GROMACS,
A. Hu, L. Pennati, S. Markidis, and I. Peng, “Enabling AI Deep Potentials for Ab Initio-quality Molecular Dynamics Simulations in GROMACS,” in2026 34th Euromicro International Conference on Parallel, Distributed, and Network-Based Processing (PDP). Los Alamitos, CA, USA: IEEE Computer Society, Mar. 2026
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.