Recognition: unknown
Evaluating the Limitations of Protein Sequence Representations for Parkinson's Disease Classification
Pith reviewed 2026-05-10 16:17 UTC · model grok-4.3
The pith
Protein primary sequences alone provide only limited discriminative power for Parkinson's disease classification.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The evaluation demonstrates that primary sequence information alone provides limited discriminative power for Parkinson's disease classification. Across amino-acid composition, k-mers, physicochemical descriptors, hybrid features, and ProtBERT embeddings, F1 scores stayed between 0.60 and 0.70 under nested stratified cross-validation. Unsupervised analyses showed no intrinsic structure aligned with class labels, and a Friedman test found no significant performance differences. Classical k-mer methods produced highly imbalanced predictions, while even the strongest configuration (ProtBERT plus MLP) yielded an F1 of 0.704 and ROC-AUC of 0.748.
What carries the argument
Nested stratified cross-validation applied to representations derived exclusively from protein primary sequences.
If this is right
- All tested sequence representations produce only moderate classification performance with substantial class overlap.
- Classical k-mer features exhibit strong bias toward positive predictions, yielding high recall but low precision.
- Unsupervised methods reveal no natural clustering that matches disease labels.
- Robust disease modeling requires biological features beyond primary sequence, such as structural or interaction data.
- The work supplies a reproducible baseline for comparing future sequence-based or multi-modal approaches.
Where Pith is reading between the lines
- Future biomarker work should integrate 3D structure, post-translational modifications, or protein interaction networks alongside sequence data.
- The same limited signal may appear when sequence-only methods are applied to other multifactorial diseases.
- Larger, cleaner protein datasets could be used to test whether the observed class overlap persists or shrinks.
Load-bearing premise
The chosen representations and dataset would detect any discriminative signal that exists in the primary sequences.
What would settle it
A primary-sequence representation that reaches F1 scores above 0.85 under identical nested stratified cross-validation on the same or a comparable Parkinson's protein dataset.
Figures





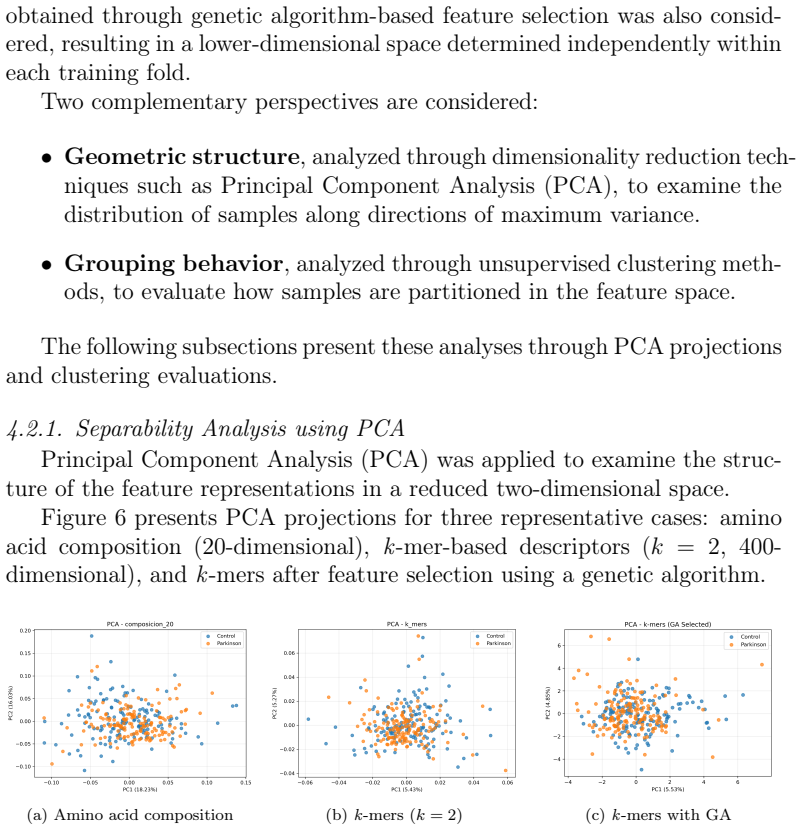
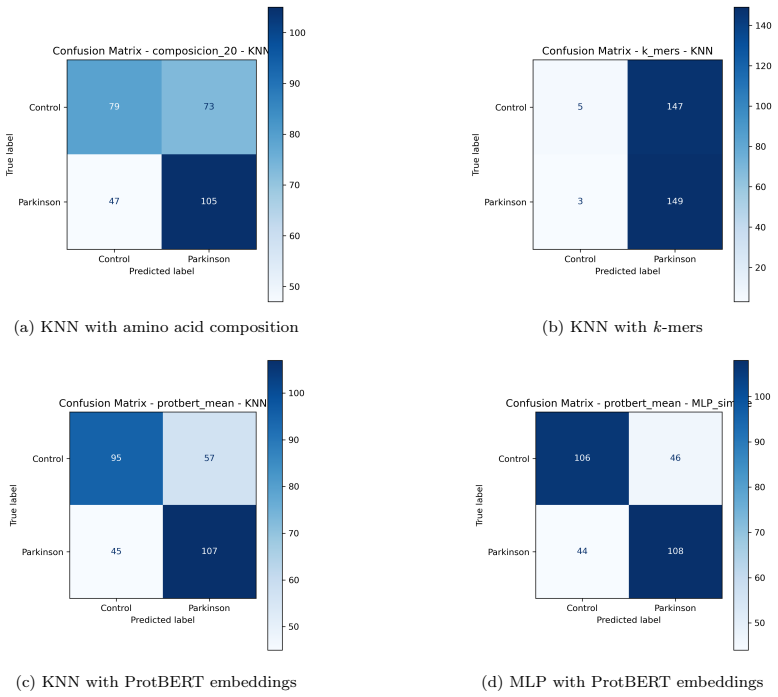


read the original abstract
The identification of reliable molecular biomarkers for Parkinson's disease remains challenging due to its multifactorial nature. Although protein sequences constitute a fundamental and widely available source of biological information, their standalone discriminative capacity for complex disease classification remains unclear. In this work, we present a controlled and leakage-free evaluation of multiple representations derived exclusively from protein primary sequences, including amino acid composition, k-mers, physicochemical descriptors, hybrid representations, and embeddings from protein language models, all assessed under a nested stratified cross-validation framework to ensure unbiased performance estimation. The best-performing configuration (ProtBERT + MLP) achieves an F1-score of 0.704 +/- 0.028 and ROC-AUC of 0.748 +/- 0.047, indicating only moderate discriminative performance. Classical representations such as k-mers reach comparable F1 values (up to approximately 0.667), but exhibit highly imbalanced behavior, with recall close to 0.98 and precision around 0.50, reflecting a strong bias toward positive predictions. Across representations, performance differences remain within a narrow range (F1 between 0.60 and 0.70), while unsupervised analyses reveal no intrinsic structure aligned with class labels, and statistical testing (Friedman test, p = 0.1749) does not indicate significant differences across models. These results demonstrate substantial overlap between classes and indicate that primary sequence information alone provides limited discriminative power for Parkinson's disease classification. This work establishes a reproducible baseline and provides empirical evidence that more informative biological features, such as structural, functional, or interaction-based descriptors, are required for robust disease modeling.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript evaluates multiple representations derived solely from protein primary sequences—including amino acid composition, k-mers, physicochemical descriptors, hybrids, and ProtBERT embeddings—for Parkinson's disease classification. Using nested stratified cross-validation, it reports moderate performance (best F1-score 0.704 ± 0.028 and ROC-AUC 0.748 ± 0.047 with ProtBERT + MLP), comparable results across classical representations (F1 up to ~0.667 but with imbalanced precision/recall), no intrinsic class structure in unsupervised analyses, and no significant differences via Friedman test (p=0.1749). The authors conclude that primary sequence information alone provides limited discriminative power and that structural, functional, or interaction-based features are needed.
Significance. If the central claim holds, the work supplies a reproducible, leakage-free baseline that quantifies the insufficiency of sequence-only approaches for PD classification and usefully directs attention toward richer biological descriptors. Strengths include the nested CV protocol, statistical testing, and focus on unbiased estimation. The result is of moderate significance for the field, as it empirically documents representational limits but would gain impact from stronger validation that the tested feature sets are representative of what primary sequences can offer.
major comments (2)
- [Abstract and Results] Abstract and Results: The claim that primary sequence information alone provides limited discriminative power is load-bearing for the paper's conclusion. It rests on the tested representations (amino-acid composition, k-mers, physicochemical descriptors, hybrids, and fixed ProtBERT embeddings) being sufficient to surface any existing signal. The manuscript does not fine-tune ProtBERT, does not specify k-mer ranges or hybrid construction, and omits comparison to stronger contemporary encoders such as ESM-2; therefore moderate performance and non-significant differences (Friedman p=0.1749) could reflect representational inadequacy rather than absence of sequence-based information.
- [Methods (data section)] Methods (data section): The representativeness assumption and absence of label noise or batch effects are central to interpreting the moderate performance as evidence of limited sequence power. Explicit details on protein dataset provenance, exact preprocessing pipeline, and any batch-effect diagnostics are required to support the claim that the evaluated sequences are a fair test of primary-sequence utility.
minor comments (2)
- [Abstract] Abstract: The statement that classical representations reach F1 values 'up to approximately 0.667' would be clearer if the full per-representation table of F1, precision, recall, and ROC-AUC values were referenced or summarized.
- The unsupervised analyses and Friedman test results are presented without citing the exact number of representations or folds used; adding these numbers would improve reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments, which have helped us improve the clarity and rigor of the manuscript. We address each major comment point by point below.
read point-by-point responses
-
Referee: [Abstract and Results] Abstract and Results: The claim that primary sequence information alone provides limited discriminative power is load-bearing for the paper's conclusion. It rests on the tested representations (amino-acid composition, k-mers, physicochemical descriptors, hybrids, and fixed ProtBERT embeddings) being sufficient to surface any existing signal. The manuscript does not fine-tune ProtBERT, does not specify k-mer ranges or hybrid construction, and omits comparison to stronger contemporary encoders such as ESM-2; therefore moderate performance and non-significant differences (Friedman p=0.1749) could reflect representational inadequacy rather than absence of sequence-based information.
Authors: We thank the referee for highlighting this key aspect of our claims. Our use of fixed ProtBERT embeddings (without fine-tuning) was a deliberate design choice to evaluate standard pre-trained representations in a strictly leakage-free nested CV setting; fine-tuning on this modest-sized, imbalanced dataset would have introduced substantial overfitting risk and complicated unbiased performance estimation. We will add an explicit justification for this choice in the revised Discussion. We will also specify the exact k-mer range (k = 1–5) and hybrid construction details (concatenation of amino-acid composition with selected physicochemical descriptors) in the Methods section. While ESM-2 is a more recent and potentially stronger encoder, the study was performed with ProtBERT as the representative protein language model available during the work; the fact that classical, non-embedding methods yield statistically indistinguishable moderate performance (F1 range 0.60–0.70, Friedman p = 0.1749) and that unsupervised visualizations show no class-aligned structure indicates that the limited discriminative power is not an artifact of any single representation family. revision: partial
-
Referee: [Methods (data section)] Methods (data section): The representativeness assumption and absence of label noise or batch effects are central to interpreting the moderate performance as evidence of limited sequence power. Explicit details on protein dataset provenance, exact preprocessing pipeline, and any batch-effect diagnostics are required to support the claim that the evaluated sequences are a fair test of primary-sequence utility.
Authors: We agree that these details are necessary for full interpretability and reproducibility. In the revised manuscript we will expand the Data and Methods sections to provide: (i) complete provenance (sequences drawn from UniProt with explicit PD-associated and control selection criteria), (ii) the full preprocessing pipeline (length filtering 50–2000 residues, duplicate removal, and handling of ambiguous residues), and (iii) batch-effect diagnostics (PCA and silhouette analysis on both raw and embedded feature spaces showing no significant source- or batch-driven clustering). These additions will be accompanied by a short supplementary note confirming the absence of detectable label noise or batch effects. revision: yes
Circularity Check
No significant circularity; direct empirical evaluation from held-out data
full rationale
The paper reports a controlled empirical evaluation of protein sequence representations (amino-acid composition, k-mers, physicochemical descriptors, hybrids, and ProtBERT embeddings) for Parkinson's disease classification. All reported metrics (F1-score 0.704, ROC-AUC 0.748, Friedman test p=0.1749) are computed via nested stratified cross-validation on held-out folds, with no derivations, fitted parameters renamed as predictions, or self-referential equations. Unsupervised analyses and statistical comparisons operate directly on the computed performance values from independent data splits. No load-bearing self-citations or ansatzes appear in the central claims; the conclusion of limited discriminative power follows from the observed moderate performance and class overlap rather than any definitional reduction.
Axiom & Free-Parameter Ledger
free parameters (1)
- MLP and classifier hyperparameters
axioms (2)
- domain assumption Nested stratified cross-validation yields unbiased performance estimates
- domain assumption The protein sequence dataset labels are accurate and representative
Reference graph
Works this paper leans on
-
[1]
P. Prajjwal, et al., Parkinson’s disease updates: Addressing the patho- physiology, risk factors, genetics, diagnosis, along with the medical and 33 surgical treatment, Annals of Medicine and Surgery 85 (10) (2023) 4887– 4902.doi:10.1097/MS9.0000000000001142
-
[2]
M. Muleiro Alvarez, et al., A comprehensive approach to Parkinson’s disease: Addressing its molecular, clinical, and therapeutic aspects, In- ternational Journal of Molecular Sciences 25 (13) (2024) 7183.doi: 10.3390/ijms25137183
-
[3]
J. S. Bogers, B. R. Bloem, J. M. Den Heijer, The etiology of Parkinson’s disease, Journal of Parkinson’s Disease 13 (2023) 1281–1288.doi:10. 3233/JPD-230250
2023
-
[4]
E. Srinivasan, G. Chandrasekhar, P. Chandrasekar, K. Anbarasu, A. S. Vickram, R. Karunakaran, R. Rajasekaran, P. S. Srikumar, Alpha- synuclein aggregation in Parkinson’s disease, Frontiers in Medicine 8 (2021) 736978.doi:10.3389/fmed.2021.736978
-
[5]
F. F. Geibl, et al., Alpha-synuclein pathology disrupts mitochondrial function, Molecular Neurodegeneration 19 (2024) 69.doi:10.1186/ s13024-024-00756-2
2024
-
[7]
Dong-Chen, et al., Signaling pathways in Parkinson’s disease, Sig- nal Transduction and Targeted Therapy 8 (2023) 73.doi:10.1038/ s41392-023-01353-3
X. Dong-Chen, et al., Signaling pathways in Parkinson’s disease, Sig- nal Transduction and Targeted Therapy 8 (2023) 73.doi:10.1038/ s41392-023-01353-3
2023
-
[8]
M. S. Khan, et al., Parkinson disease signaling pathways, International Journal of Molecular Sciences 26 (13) (2025) 6416.doi:10.3390/ ijms26136416
2025
-
[9]
J. Blesa, et al., Oxidative stress and Parkinson’s disease, Frontiers in Neuroanatomy 9 (2015) 91.doi:10.3389/fnana.2015.00091
-
[10]
Zarkali, et al., Neuroimaging and fluid biomarkers in Parkin- son’s disease, Nature Communications 15 (2024) 5661.doi:10.1038/ s41467-024-49949-9
A. Zarkali, et al., Neuroimaging and fluid biomarkers in Parkin- son’s disease, Nature Communications 15 (2024) 5661.doi:10.1038/ s41467-024-49949-9. 34
2024
-
[11]
J. Mei, et al., Machine learning for Parkinson’s disease diagnosis, Fron- tiers in Aging Neuroscience 13 (2021) 633752.doi:10.3389/fnagi. 2021.633752
-
[12]
A. Díaz-Ramírez, J. Díaz-Escobar, V. Quintero-Rosas, R. Moncada- Sánchez, Classification of fall events in the elderly using a thermal sensor and machine learning techniques, Computación y Sistemas 28 (4) (2024) 1773–1787.doi:10.13053/cys-28-4-4809
-
[13]
H. Rabie, M. A. Akhloufi, Machine learning and deep learning for Parkinson’s disease detection, Discover Artificial Intelligence 5 (2025) 24.doi:10.1007/s44163-025-00241-9
-
[14]
S. Seo, M. Oh, Y. Park, S. Kim, Deepfam: deep learning based alignment-free method for protein family modeling and pre- diction, Bioinformatics 34 (13) (2018) i254–i262.doi:10.1093/ bioinformatics/bty275
2018
-
[15]
J. J. Almagro Armenteros, C. K. Sønderby, S. K. Sønderby, H. Nielsen, O. Winther, Deeploc: prediction of protein subcellular localization using deep learning, Bioinformatics 33 (21) (2017) 3387–3395.doi:10.1093/ bioinformatics/btx431
2017
-
[16]
E. Asgari, M. R. Mofrad, Continuous distributed representation of bio- logical sequences for deep proteomics and genomics, PLoS ONE 10 (11) (2015) e0141287.doi:10.1371/journal.pone.0141287
-
[17]
Rao, et al., Evaluating protein transfer learning with tape, in: Ad- vances in Neural Information Processing Systems, 2019
R. Rao, et al., Evaluating protein transfer learning with tape, in: Ad- vances in Neural Information Processing Systems, 2019
2019
-
[18]
ProtTrans : Toward understanding the language of life through self-supervised learning
A. Elnaggar, M. Heinzinger, C. Dallago, G. Rehawi, Y. Wang, L. Jones, T. Gibbs, T. Feher, C. Angerer, D. Bhowmik, B. Rost, Prottrans: Toward cracking the language of life’s code through self-supervised deep learning and high performance computing, IEEE Transactions on Pattern Analysis and Machine Intelligence 44 (10) (2022) 7112–7127. doi:10.1109/TPAMI.20...
-
[19]
A. Rives, et al., Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, Proceedings of the National Academy of Sciences 118 (15) (2021) e2016239118.doi: 10.1073/pnas.2016239118. 35
-
[20]
K.-C. Chou, Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology, Journal of Theoretical Biology 273 (2011) 236–247.doi:10.1016/j.jtbi.2010.12.024
-
[21]
A. Zielezinski, et al., Alignment-free sequence comparison, Genome Bi- ology 18 (2017) 186.doi:10.1186/s13059-017-1319-7
-
[22]
Z. Lin, et al., Evolutionary-scale prediction of atomic-level protein struc- ture with a language model, Science 379 (6637) (2023) 1123–1130. doi:10.1126/science.ade2574
-
[23]
P. Radivojac, W. T. Clark, T. Oron, A. M. Schnoes, T. Wittkop, A. Sokolov, K. Graim, C. Funk, K. Verspoor, A. Ben-Hur, G. Pandey, J. M. Yunes, A. S. Talwalkar, S. Repo, M. L. Souza, D. Piovesan, R. Casadio, Z. Wang, J. Cheng, H. Fang, J. Gough, P. Koskinen, P. Törönen, J. Nokso-Koivisto, L. Holm, D. Cozzetto, D. W. Buchan, K.Bryson, D.T.Jones, B.Limaye, H...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.