Recognition: unknown
Interpretable DNA Sequence Classification via Dynamic Feature Generation in Decision Trees
Pith reviewed 2026-05-10 15:19 UTC · model grok-4.3
The pith
DEFT uses large language models to generate dynamic high-level features during decision tree construction for DNA sequences.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
DEFT leverages large language models to propose biologically-informed features tailored to the local sequence distributions at each node and to iteratively refine them with a reflection mechanism, enabling axis-aligned decision trees to handle high-level sequence features for interpretable DNA analysis.
What carries the argument
DEFT's adaptive feature generation, in which large language models propose and refine sequence features matched to local node data during tree growth.
If this is right
- Decision trees gain expressivity to capture complex DNA patterns without requiring prohibitive depth.
- The discovered features remain human-readable while delivering strong predictive performance.
- The same process applies across multiple genomic classification tasks.
- Models avoid the opacity of deep neural networks while improving on raw-feature trees.
Where Pith is reading between the lines
- The method could extend to protein or RNA sequences if the language model step generalizes.
- Domain experts could directly edit or validate the LLM-proposed features to inject prior knowledge.
- Performance may vary sharply with the quality of the underlying large language model.
Load-bearing premise
Large language models can reliably propose biologically-informed features tailored to local sequence distributions at each node and that the reflection mechanism will iteratively refine them into useful splits.
What would settle it
If, on standard genomic benchmarks, DEFT-built trees show no gain in accuracy or interpretability over ordinary decision trees, or if biologists judge the generated features as uninformative, the central claim would be refuted.
Figures







read the original abstract
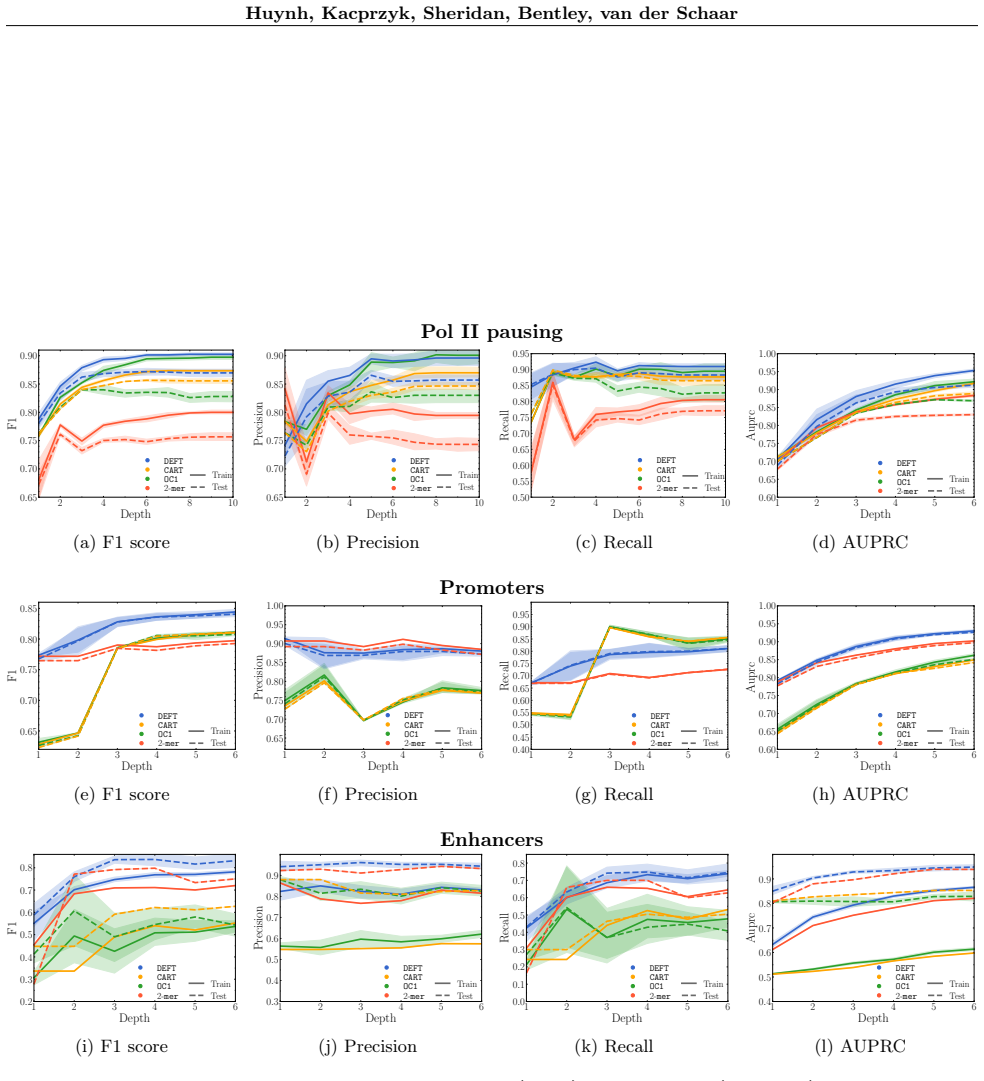
The analysis of DNA sequences has become critical in numerous fields, from evolutionary biology to understanding gene regulation and disease mechanisms. While deep neural networks can achieve remarkable predictive performance, they typically operate as black boxes. Contrasting these black boxes, axis-aligned decision trees offer a promising direction for interpretable DNA sequence analysis, yet they suffer from a fundamental limitation: considering individual raw features in isolation at each split limits their expressivity, which results in prohibitive tree depths that hinder both interpretability and generalization performance. We address this challenge by introducing DEFT, a novel framework that adaptively generates high-level sequence features during tree construction. DEFT leverages large language models to propose biologically-informed features tailored to the local sequence distributions at each node and to iteratively refine them with a reflection mechanism. Empirically, we demonstrate that DEFT discovers human-interpretable and highly predictive sequence features across a diverse range of genomic tasks.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces DEFT, a framework for interpretable DNA sequence classification that augments axis-aligned decision trees by using large language models to dynamically propose and refine biologically-informed, high-level sequence features at each node based on local distributions. These features are intended to increase expressivity beyond raw k-mers, yielding shallower trees that remain human-interpretable while achieving strong predictive performance across genomic tasks.
Significance. If the empirical claims are substantiated with rigorous validation, the work could provide a practical route to interpretable models in genomics that avoid the depth and opacity problems of standard decision trees while leveraging LLM capabilities for feature generation. This would be a meaningful contribution to the intersection of interpretable ML and bioinformatics, particularly if the method generalizes beyond the reported tasks.
major comments (3)
- [Empirical evaluation / Results] The central empirical claim (that DEFT discovers human-interpretable and highly predictive features) is not supported by any reported metrics, baselines, ablation studies, dataset sizes, or statistical comparisons in the abstract or described evaluation. Without these, it is impossible to assess whether the LLM-generated features outperform standard k-mer or one-hot encodings or whether the reflection mechanism improves accuracy or reduces depth.
- [Method description (feature proposal and reflection)] The reliability of LLM-proposed features is load-bearing for both the interpretability and performance claims, yet no grounding mechanism, hallucination detection, expert validation, motif enrichment analysis, or comparison to curated biological databases is described. This leaves open the possibility that proposed features are ungrounded or unstable across runs.
- [Algorithm and experimental setup] No quantitative analysis is provided on how the reflection loop affects tree depth, convergence, or generalization relative to gradient-boosted trees or standard CART with k-mers. This is required to substantiate the claim that the approach solves the expressivity limitation of axis-aligned splits.
minor comments (1)
- [Abstract] The abstract would benefit from a concise statement of the datasets, tasks, and key quantitative results to allow readers to immediately gauge the strength of the empirical demonstration.
Simulated Author's Rebuttal
We thank the referee for the thoughtful and constructive feedback. We appreciate the identification of areas where the presentation of empirical results, method validation, and algorithmic analysis can be strengthened. We address each major comment below and will incorporate revisions to improve the manuscript.
read point-by-point responses
-
Referee: [Empirical evaluation / Results] The central empirical claim (that DEFT discovers human-interpretable and highly predictive features) is not supported by any reported metrics, baselines, ablation studies, dataset sizes, or statistical comparisons in the abstract or described evaluation. Without these, it is impossible to assess whether the LLM-generated features outperform standard k-mer or one-hot encodings or whether the reflection mechanism improves accuracy or reduces depth.
Authors: We acknowledge that the abstract and high-level evaluation description do not include specific numerical metrics or explicit baseline comparisons, which limits immediate assessment of the claims. The full manuscript reports experiments across multiple genomic tasks showing predictive performance and shallower trees, but these details are not sufficiently quantified or compared in the current text. In the revised manuscript, we will update the abstract with key results (e.g., accuracy gains and depth reductions), add a results table with dataset sizes, performance metrics against k-mer CART, one-hot baselines, and gradient-boosted trees, include ablation studies isolating the reflection mechanism, and report statistical comparisons such as paired t-tests or Wilcoxon tests. revision: yes
-
Referee: [Method description (feature proposal and reflection)] The reliability of LLM-proposed features is load-bearing for both the interpretability and performance claims, yet no grounding mechanism, hallucination detection, expert validation, motif enrichment analysis, or comparison to curated biological databases is described. This leaves open the possibility that proposed features are ungrounded or unstable across runs.
Authors: We agree that explicit validation of the LLM-proposed features is essential to support both interpretability and reliability claims. The manuscript relies on the reflection mechanism for iterative refinement based on local data but does not describe additional grounding steps. We will revise the methods section to add motif enrichment analysis (e.g., using tools like MEME or HOMER), comparisons against curated databases such as JASPAR or TRANSFAC, stability metrics across multiple independent LLM runs with varied temperatures, and, where possible, a small-scale expert biologist review of a subset of proposed features to detect potential hallucinations or ungrounded proposals. revision: yes
-
Referee: [Algorithm and experimental setup] No quantitative analysis is provided on how the reflection loop affects tree depth, convergence, or generalization relative to gradient-boosted trees or standard CART with k-mers. This is required to substantiate the claim that the approach solves the expressivity limitation of axis-aligned splits.
Authors: The referee is correct that the current manuscript describes the reflection loop at a high level without quantitative ablations on its effects. While the algorithm section explains the dynamic feature generation and reflection process, it lacks direct measurements of impact on depth, convergence, or generalization. In the revision, we will add dedicated experimental subsections and figures quantifying the reflection loop's contribution (e.g., tree depth and accuracy with/without reflection), along with head-to-head comparisons against standard CART using k-mers and gradient-boosted trees, including metrics on generalization to held-out sequences and convergence behavior during tree construction. revision: yes
Circularity Check
No circularity: DEFT method relies on external LLM components without self-referential reductions or fitted predictions.
full rationale
The paper introduces DEFT as an empirical framework that uses large language models to generate and refine sequence features during decision tree construction for DNA classification. No mathematical derivations, equations, or parameter-fitting procedures are described that could reduce a claimed result to its own inputs by construction. The central claims rest on empirical demonstrations across genomic tasks rather than any self-definitional loop, uniqueness theorem imported from prior self-work, or renaming of known results. The LLM proposal and reflection steps are presented as external mechanisms, not derived from the paper's own fitted quantities or citations that collapse the argument. This is a standard non-circular empirical ML contribution.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Veloso, A., Kirkconnell, K., Magnuson, B., Biewen, B., Paulsen, M., Wilson, T., and Ljungman, M
PMLR. Veloso, A., Kirkconnell, K., Magnuson, B., Biewen, B., Paulsen, M., Wilson, T., and Ljungman, M. (2014). Rate of elongation by rna polymerase ii is associated with specific gene features and epigenetic modifica- tions.Genome Res, 24:896–905. Verwer, S. and Zhang, Y. (2019). Learning optimal classification trees using a binary linear program formulat...
2014
-
[2]
Springer. Wang, R., Wang, Z., Wang, J., and Li, S. (2019). Splicefinder: ab initio prediction of splice sites using convolutional neural network.BMC bioinformatics, 20:1–13. Wang, R., Zelikman, E., Poesia, G., Pu, Y., Haber, N., and Goodman, N. D. (2023). Hypothesis search: Inductive reasoning with language models.arXiv preprint arXiv:2309.05660. Yang, C....
-
[3]
under the Apache-2.0 license. It involves classifying251bp genomic sequences as either non-TATA promoters (positive class; sequences spanning -200bp to +50bp around a transcription start site lacking a TATA-box) or non-promoter regions (negative class; random251bp fragments from human gene regions located after the first exons). Intuitively, this promoter...
2016
-
[4]
thetask context C: describes the input space, the label, the characteristics of the dataset, and the tree induction task
-
[5]
ra ti on ale
thenode contextS nl v,T : lists the sequence of splitting conditions from the root node to the current nodev 3.interpretability instructions: prevents composite features combining multiple mechanisms 4.task-specific instructions: for example, instructions for exploration and exploitation in reflection In addition to that, the reflection prompts contain in...
-
[6]
ra ti on ale
Name : clear and d e s c r i p t i v e . Return in the f ol lo win g JSON format : { " ra ti on ale " : " y o u r _ r a t i o n a l e " , " d e s c r i p t i o n " : " y o u r _ f e a t u r e _ d e s c r i p t i o n " , " name " : " y o u r _ f e a t u r e _ n a m e " } Only return the JSON object , with no a d d i t i o n a l text . Do not include " json...
-
[7]
ra ti on ale
Name : clear and d e s c r i p t i v e . Return in the f ol lo win g JSON format : { " ra ti on ale " : " y o u r _ r a t i o n a l e " , " d e s c r i p t i o n " : " y o u r _ f e a t u r e _ d e s c r i p t i o n " , " name " : " y o u r _ f e a t u r e _ n a m e " } Only return the JSON object , with no a d d i t i o n a l text . Do not include " json...
1994
-
[8]
Faithfulness of rationale to feature and task: How accurately does the rationale describe what the feature’s code computes and why that computation is relevant for Pol II pausing?
-
[9]
The expert also found that DEFT largely proposed features plausibly aligned with determinants of pausing and transcriptional mechanics (e.g
Biological interpretability: To what extent does the proposed mechanism correspond to a single, coherent biological phenomenon and appear biologically plausible? Results.The expert judged that the rationales almost always correctly described the executable feature and articulated a task-relevant mechanism, yielding a score of4.89 ± 0.46out of5on faithfuln...
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.