Recognition: unknown
A Systematic Survey and Benchmark of Deep Learning for Molecular Property Prediction in the Foundation Model Era
Pith reviewed 2026-05-10 08:39 UTC · model grok-4.3
The pith
A systematic survey and benchmark of four deep learning paradigms for molecular property prediction that organizes the field, critiques current data practices, and outlines three future directions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Current benchmark practices suffer from inconsistent stereochemistry handling, heterogeneous assay sources, and reproducibility limitations under random or poorly defined splits, motivating modernization toward transparent, time- and scaffold-aware methodologies.
Load-bearing premise
That the surveyed datasets and identified challenges sufficiently represent the full range of real-world molecular prediction tasks and that the three proposed directions (physics-aware learning, uncertainty calibration, multimodal benchmarks) will measurably improve model reliability without introducing new unaddressed biases.
Figures




read the original abstract
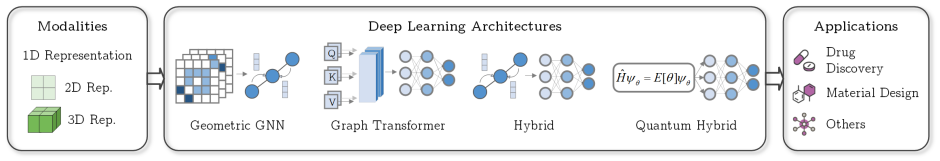
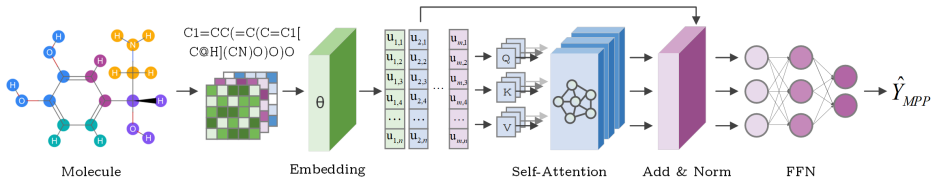
Molecular property prediction integrates quantum chemistry, cheminformatics, and deep learning to connect molecular structure with physicochemical and biological behavior. This survey traces four complementary paradigms, including Quantum, Descriptor Machine Learning, Geometric Deep Learning, and Foundation Models, and outlines a unified taxonomy linking molecular representations, model architectures, and interdisciplinary applications. Benchmark analyses integrate evidence from both widely used datasets and datasets reflecting industry perspectives, encompassing quantum, physicochemical, physiological, and biophysical domains. The survey examines current standards in data curation, splitting strategies, and evaluation protocols, highlighting challenges including inconsistent stereochemistry, heterogeneous assay sources, and reproducibility limitations under random or poorly defined splits. These observations motivate the modernization of benchmark design toward more transparent, time- and scaffold-aware methodologies. We further propose three forward-looking directions: (i) physics-aware learning embedding quantum consistency, (ii) uncertainty-calibrated foundation models for trustworthy inference, and (iii) realistic multimodal benchmark ecosystems integrating computational and experimental data. Repository: https://github.com/Zongru-Li/Survey-and-Benchmarks-of-DL-for-Molecular-Property-Prediction-in-the-Foundation-Model-Era.
Editorial analysis
A structured set of objections, weighed in public.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
K. T. Schütt, P.-J. Kindermans, H. E. Sauceda, S. Chmiela, A. Tkatchenko, and K.-R. Müller. Schnet: a continuous-filter convolutional neural network for modeling quantum interactions. InProceedings of the 31st International Conference on Neural Information Processing Systems, NIPS’17, page 992–1002, Red Hook, NY , USA, 2017. Curran Associates Inc
2017
-
[2]
Schoenholz, Patrick F
Justin Gilmer, Samuel S. Schoenholz, Patrick F. Riley, Oriol Vinyals, and George E. Dahl. Neural message passing for quantum chemistry. InProceedings of the 34th International Conference on Machine Learning - Volume 70, ICML’17, page 1263–1272. JMLR.org, 2017
2017
-
[3]
Anatole von Lilienfeld, Klaus-Robert Müller, and Alexandre Tkatchenko
O. Anatole von Lilienfeld, Klaus-Robert Müller, and Alexandre Tkatchenko. Exploring chemical compound space with quantum-based machine learning.Nature Reviews Chemistry, 4(7):347–358, 2020
2020
-
[4]
Applications of machine learning in drug discovery and development.Nature Reviews Drug Discovery, 18(6):463–477, 2019
Jessica Vamathevan, Dominic Clark, Paul Czodrowski, Ian Dunham, Edgardo Ferran, George Lee, Bin Li, Anant Madabhushi, Parantu Shah, Michaela Spitzer, and Shanrong Zhao. Applications of machine learning in drug discovery and development.Nature Reviews Drug Discovery, 18(6):463–477, 2019
2019
-
[5]
A compact review of molecular property prediction with graph neural networks.Drug Discovery Today: Technologies, 37:1–12, 2020
Oliver Wieder, Stefan Kohlbacher, Mélaine Kuenemann, Arthur Garon, Pierre Ducrot, Thomas Seidel, and Thierry Langer. A compact review of molecular property prediction with graph neural networks.Drug Discovery Today: Technologies, 37:1–12, 2020
2020
-
[6]
A perspective on foundation models in chemistry.JACS Au, 5(4):1499–1518, 2025
Junyoung Choi, Gunwook Nam, Jaesik Choi, and Yousung Jung. A perspective on foundation models in chemistry.JACS Au, 5(4):1499–1518, 2025
2025
-
[7]
Advancements in molecular property prediction: A survey of single and multimodal approaches.Archives of Computational Methods in Engineering, pages 1–31, 2025
Tanya Liyaqat, Tanvir Ahmad, and Chandni Saxena. Advancements in molecular property prediction: A survey of single and multimodal approaches.Archives of Computational Methods in Engineering, pages 1–31, 2025
2025
-
[8]
Calculation of local excitations in large systems by embedding wave-function theory in density-functional theory.Physical Chemistry Chemical Physics, 10(35):5353–5362, 2008
André Severo Pereira Gomes, Christoph R Jacob, and Lucas Visscher. Calculation of local excitations in large systems by embedding wave-function theory in density-functional theory.Physical Chemistry Chemical Physics, 10(35):5353–5362, 2008
2008
-
[9]
Physnet: A neural network for predicting energies, forces, dipole moments, and partial charges.Journal of Chemical Theory and Computation, 15(6):3678–3693, 2019
Oliver T Unke and Markus Meuwly. Physnet: A neural network for predicting energies, forces, dipole moments, and partial charges.Journal of Chemical Theory and Computation, 15(6):3678–3693, 2019
2019
-
[10]
The performance of density functional and wavefunction- based methods for 2d and 3d structures of au10.Journal of Computational Chemistry, 34(23):1975–1981, 2013
Daniel A Götz, Rolf Schäfer, and Peter Schwerdtfeger. The performance of density functional and wavefunction- based methods for 2d and 3d structures of au10.Journal of Computational Chemistry, 34(23):1975–1981, 2013
1975
-
[11]
Linear-scaling quantum mechanical calculations of biological molecules: The divide-and-conquer approach.Computational Materials Science, 12(3):259–277, 1998
Tai-Sung Lee, James P Lewis, and Weitao Yang. Linear-scaling quantum mechanical calculations of biological molecules: The divide-and-conquer approach.Computational Materials Science, 12(3):259–277, 1998
1998
-
[12]
Large-scale molecular simulations of hypervelocity impact of materials.Procedia Engineering, 58:167–176, 2013
Andres Jaramillo-Botero, Qi An, Patrick L Theofanis, and William A Goddard III. Large-scale molecular simulations of hypervelocity impact of materials.Procedia Engineering, 58:167–176, 2013
2013
-
[13]
Extended-connectivity fingerprints.Journal of chemical information and modeling, 50(5):742–754, 2010
David Rogers and Mathew Hahn. Extended-connectivity fingerprints.Journal of chemical information and modeling, 50(5):742–754, 2010
2010
-
[14]
Ling Xue, Jeffrey W Godden, Florence L Stahura, and Jürgen Bajorath. Design and evaluation of a molecular fingerprint involving the transformation of property descriptor values into a binary classification scheme.Journal of chemical information and computer sciences, 43(4):1151–1157, 2003
2003
-
[15]
Reconstruction of lossless molecular representations from fingerprints.Journal of cheminformatics, 15(1):26, 2023
Umit V Ucak, Islambek Ashyrmamatov, and Juyong Lee. Reconstruction of lossless molecular representations from fingerprints.Journal of cheminformatics, 15(1):26, 2023. 21
2023
-
[16]
Geometry-enhanced molecular representation learning for property prediction.Nature Machine Intelligence, 4(2):127–134, 2022
Xiaomin Fang, Lihang Liu, Jieqiong Lei, Donglong He, Shanzhuo Zhang, Jingbo Zhou, Fan Wang, Hua Wu, and Haifeng Wang. Geometry-enhanced molecular representation learning for property prediction.Nature Machine Intelligence, 4(2):127–134, 2022
2022
-
[17]
Se (3)-transformers: 3d roto-translation equivariant attention networks.Advances in neural information processing systems, 33:1970–1981, 2020
Fabian Fuchs, Daniel Worrall, V olker Fischer, and Max Welling. Se (3)-transformers: 3d roto-translation equivariant attention networks.Advances in neural information processing systems, 33:1970–1981, 2020
1970
-
[18]
Directed message passing based on attention for prediction of molecular properties
Gong Chen and Yvon Maday. Directed message passing based on attention for prediction of molecular properties. Computational Materials Science, 229:112443, 2023
2023
-
[19]
Generalizing from a few examples: A survey on few-shot learning.ACM computing surveys (csur), 53(3):1–34, 2020
Yaqing Wang, Quanming Yao, James T Kwok, and Lionel M Ni. Generalizing from a few examples: A survey on few-shot learning.ACM computing surveys (csur), 53(3):1–34, 2020
2020
-
[20]
Fs-mol: A few-shot learning dataset of molecules
Megan Stanley, John F Bronskill, Krzysztof Maziarz, Hubert Misztela, Jessica Lanini, Marwin Segler, Nadine Schneider, and Marc Brockschmidt. Fs-mol: A few-shot learning dataset of molecules. InThirty-fifth Conference on Neural Information Processing Systems Datasets and Benchmarks Track (Round 2), 2021
2021
-
[21]
Nawaf Alampara, Mara Schilling-Wilhelmi, Martiño Ríos-García, Indrajeet Mandal, Pranav Khetarpal, Har- gun Singh Grover, N. M. Anoop Krishnan, and Kevin Maik Jablonka. Probing the limitations of multimodal language models for chemistry and materials research.Nature Computational Science, 5(10):952–961, Oct 2025
2025
-
[22]
Evaluating large language models on multimodal chemistry olympiad exams.Communications Chemistry, 8(1):402, Dec 2025
Yiming Cui, Xin Yao, Yuxuan Qin, Xin Li, Shijin Wang, and Guoping Hu. Evaluating large language models on multimodal chemistry olympiad exams.Communications Chemistry, 8(1):402, Dec 2025
2025
-
[23]
Ash, Cas Wognum, Raquel Rodríguez-Pérez, Matteo Aldeghi, Alan C
Jeremy R. Ash, Cas Wognum, Raquel Rodríguez-Pérez, Matteo Aldeghi, Alan C. Cheng, Djork-Arné Clevert, Ola Engkvist, Cheng Fang, Daniel J. Price, Jacqueline M. Hughes-Oliver, and W. Patrick Walters. Practically significant method comparison protocols for machine learning in small molecule drug discovery.Journal of Chemical Information and Modeling, 65(18):...
2025
-
[24]
DeepTox: Toxicity Prediction using Deep Learning.Frontiers in Environmental Science, 3, 2016
Andreas Mayr, Günter Klambauer, Thomas Unterthiner, and Sepp Hochreiter. DeepTox: Toxicity Prediction using Deep Learning.Frontiers in Environmental Science, 3, 2016
2016
-
[25]
Fate-tox: fragment attention transformer for E(3)-equivariant multi-organ toxicity prediction.Journal of Cheminformatics, 17(1):74, May 2025
Sumin Ha, Dongmin Bang, and Sun Kim. Fate-tox: fragment attention transformer for E(3)-equivariant multi-organ toxicity prediction.Journal of Cheminformatics, 17(1):74, May 2025
2025
-
[26]
DeepDTA: deep drug–target binding affinity prediction
Hakime Öztürk, Arzucan Özgür, and Elif Ozkirimli. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics, 34(17):i821–i829, September 2018
2018
-
[27]
Seyone Chithrananda, Gabriel Grand, Bharath Ramsun- dar, et al
Walid Ahmad, Elana Simon, Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. Chemberta-2: Towards chemical foundation models.arXiv preprint arXiv:2209.01712, 2022
-
[28]
Molecular representation learning with language models and domain-relevant auxiliary tasks, 2020
Benedek Fabian, Thomas Edlich, Héléna Gaspar, Marwin Segler, Joshua Meyers, Marco Fiscato, and Mohamed Ahmed. Molecular representation learning with language models and domain-relevant auxiliary tasks, 2020
2020
-
[29]
Rouyi Wang, Shujun Yi, Guoqiang Shan, and Lingyan Zhu. Integration of transfer learning and multitask learning to predict the potential of per/polyfluoroalkyl substances in activating multiple nuclear receptors associated with hepatic lipotoxicity.Environmental Science & Technology, 59(44):23727–23738, 2025
2025
-
[30]
Mole: a foundation model for molecular graphs using disentangled attention.Nature Communications, 15(1):9431, 2024
Oscar Méndez-Lucio, Christos A Nicolaou, and Berton Earnshaw. Mole: a foundation model for molecular graphs using disentangled attention.Nature Communications, 15(1):9431, 2024
2024
-
[31]
Uni-mol: A universal 3d molecular representation learning framework
Gengmo Zhou, Zhifeng Gao, Qiankun Ding, Hang Zheng, Hongteng Xu, Zhewei Wei, Linfeng Zhang, and Guolin Ke. Uni-mol: A universal 3d molecular representation learning framework. InThe Eleventh International Conference on Learning Representations, 2023
2023
-
[32]
Hoffman, C
Vijil Chenthamarakshan, Samuel C. Hoffman, C. David Owen, Petra Lukacik, Claire Strain-Damerell, Daren Fearon, Tika R. Malla, Anthony Tumber, Christopher J. Schofield, Helen M.E. Duyvesteyn, Wanwisa Dejnirattisai, Loic Carrique, Thomas S. Walter, Gavin R. Screaton, Tetiana Matviiuk, Aleksandra Mojsilovic, Jason Crain, Martin A. Walsh, David I. Stuart, and...
2023
-
[33]
ADMET-AI: a machine learning ADMET platform for evaluation of large-scale chemical libraries
Kyle Swanson, Parker Walther, Jeremy Leitz, Souhrid Mukherjee, Joseph C Wu, Rabindra V Shivnaraine, and James Zou. ADMET-AI: a machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics, 40(7):btae416, July 2024
2024
-
[34]
Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S
Zhenqin Wu, Bharath Ramsundar, Evan N. Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S. Pappu, Karl Leswing, and Vijay Pande. Moleculenet: a benchmark for molecular machine learning.Chem. Sci., 9:513–530, 2018
2018
-
[35]
Schoenholz, Muratahan Aykol, Gowoon Cheon, and Ekin Dogus Cubuk
Amil Merchant, Simon Batzner, Samuel S. Schoenholz, Muratahan Aykol, Gowoon Cheon, and Ekin Dogus Cubuk. Scaling deep learning for materials discovery.Nature, 624(7990):80–85, December 2023
2023
-
[36]
A predictive machine learning force-field framework for liquid electrolyte development.Nature Machine Intelligence, 7(4):543–552, April 2025
Sheng Gong, Yumin Zhang, Zhenliang Mu, Zhichen Pu, Hongyi Wang, Xu Han, Zhiao Yu, Mengyi Chen, Tianze Zheng, Zhi Wang, Lifei Chen, Zhenze Yang, Xiaojie Wu, Shaochen Shi, Weihao Gao, Wen Yan, and Liang Xiang. A predictive machine learning force-field framework for liquid electrolyte development.Nature Machine Intelligence, 7(4):543–552, April 2025
2025
-
[37]
Uni-electrolyte: An artificial intelligence platform for designing electrolyte molecules for rechargeable batteries.Angewandte Chemie International Edition, 64(30):e202503105, 2025
Xiang Chen, Mingkang Liu, Shiqiu Yin, Yu-Chen Gao, Nan Yao, and Qiang Zhang. Uni-electrolyte: An artificial intelligence platform for designing electrolyte molecules for rechargeable batteries.Angewandte Chemie International Edition, 64(30):e202503105, 2025
2025
-
[38]
Do transformers really perform badly for graph representation? InThirty-Fifth Conference on Neural Information Processing Systems, 2021
Chengxuan Ying, Tianle Cai, Shengjie Luo, Shuxin Zheng, Guolin Ke, Di He, Yanming Shen, and Tie-Yan Liu. Do transformers really perform badly for graph representation? InThirty-Fifth Conference on Neural Information Processing Systems, 2021
2021
-
[39]
Graphmae: Self- supervised masked graph autoencoders
Zhenyu Hou, Xiao Liu, Yukuo Cen, Yuxiao Dong, Hongxia Yang, Chunjie Wang, and Jie Tang. Graphmae: Self- supervised masked graph autoencoders. InProceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, pages 594–604, 2022
2022
-
[40]
Chemberta: Large-scale self-supervised pretrain- ing for molecular property prediction, 2020
Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. Chemberta: Large-scale self-supervised pretrain- ing for molecular property prediction, 2020
2020
-
[41]
Molecular contrastive learning of representations via graph neural networks.Nature Machine Intelligence, 4(3):279–287, 2022
Yuyang Wang, Jianren Wang, Zhonglin Cao, and Amir Barati Farimani. Molecular contrastive learning of representations via graph neural networks.Nature Machine Intelligence, 4(3):279–287, 2022
2022
-
[42]
Nadkarni, Benjamin S
Kexin Huang, Payal Chandak, Qianwen Wang, Shreyas Havaldar, Akhil Vaid, Jure Leskovec, Girish N. Nadkarni, Benjamin S. Glicksberg, Nils Gehlenborg, and Marinka Zitnik. A foundation model for clinician-centered drug repurposing.Nature Medicine, 30(12):3601–3613, December 2024
2024
-
[43]
Allegro-fm: Toward an equivariant foundation model for exascale molecular dynamics simulations.The Journal of Physical Chemistry Letters, 16:6637–6644, 2025
Ken-ichi Nomura, Shinnosuke Hattori, Satoshi Ohmura, Ikumi Kanemasu, Kohei Shimamura, Nabankur Das- gupta, Aiichiro Nakano, Rajiv K Kalia, and Priya Vashishta. Allegro-fm: Toward an equivariant foundation model for exascale molecular dynamics simulations.The Journal of Physical Chemistry Letters, 16:6637–6644, 2025
2025
-
[44]
Smiles-bert: Large scale unsupervised pre-training for molecular property prediction
Sheng Wang, Yuzhi Guo, Yuhong Wang, Hongmao Sun, and Junzhou Huang. Smiles-bert: Large scale unsupervised pre-training for molecular property prediction. InProceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, BCB ’19, page 429–436, New York, NY , USA, 2019. Association for Computing Machinery
2019
-
[45]
Transformers for molecular property prediction: Domain adaptation efficiently improves performance, 2025
Afnan Sultan, Max Rausch-Dupont, Shahrukh Khan, Olga Kalinina, Dietrich Klakow, and Andrea V olkamer. Transformers for molecular property prediction: Domain adaptation efficiently improves performance, 2025
2025
-
[46]
Mol2vec: Unsupervised machine learning approach with chemical intuition.Journal of Chemical Information and Modeling, 58(1):27–35, 2018
Sabrina Jaeger, Simone Fulle, and Samo Turk. Mol2vec: Unsupervised machine learning approach with chemical intuition.Journal of Chemical Information and Modeling, 58(1):27–35, 2018. PMID: 29268609
2018
-
[47]
Bemis and Mark A
Guy W. Bemis and Mark A. Murcko. The properties of known drugs. 1. Molecular frameworks.J. Med. Chem., 39(15):2887–2893, 1996
1996
-
[48]
Unsupervised data base clustering based on Daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets.J
Darko Butina. Unsupervised data base clustering based on Daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets.J. Chem. Inf. Comput. Sci., 39(4):747–750, 1999. 23
1999
-
[49]
Ballester
Qianrong Guo, Saiveth Hernandez-Hernandez, and Pedro J. Ballester. Scaffold splits overestimate virtual screening performance. InArtificial Neural Networks and Machine Learning – ICANN 2024, volume 15025 of Lecture Notes in Computer Science, pages 58–72. Springer, 2024
2024
-
[50]
Ballester
Qianrong Guo, Saiveth Hernandez-Hernandez, and Pedro J. Ballester. UMAP-based clustering split for rigorous evaluation of AI models for virtual screening on cancer cell lines.J. Cheminform., 17(1):94, 2025
2025
-
[51]
Sheridan
Robert P. Sheridan. Time-split cross-validation as a method for estimating the goodness of prospective prediction. J. Chem. Inf. Model., 53(4):783–790, 2013
2013
-
[52]
Patrick Walters
W. Patrick Walters. Some thoughts on splitting chemical datasets. Practical Cheminformatics Blog, https: //practicalcheminformatics.blogspot.com/2024/11/some-thoughts-on-splitting- chemical.html, 2024. Accessed: 2026
2024
-
[53]
Uncertainty quantification and propagation in atomistic machine learning.Reviews in Chemical Engineering, 41(4):333–357, 2025
Jin Dai, Santosh Adhikari, and Mingjian Wen. Uncertainty quantification and propagation in atomistic machine learning.Reviews in Chemical Engineering, 41(4):333–357, 2025
2025
-
[54]
AkshatKumar Nigam, Robert Pollice, Matthew F. D. Hurley, Riley J. Hickman, Matteo Aldeghi, Naruki Yoshikawa, Seyone Chithrananda, Vincent A. V oelz, and Alán Aspuru-Guzik. Assigning confidence to molecular property prediction.Expert Opinion on Drug Discovery, 16(9):1009–1023, September 2021
2021
-
[55]
Lior Hirschfeld, Kyle Swanson, Kevin Yang, Regina Barzilay, and Connor W. Coley. Uncertainty quantification using neural networks for molecular property prediction.Journal of Chemical Information and Modeling, 60(8):3770–3780, August 2020
2020
-
[56]
Grambow, Barbara Pernici, Yi-Pei Li, and William H
Gabriele Scalia, Colin A. Grambow, Barbara Pernici, Yi-Pei Li, and William H. Green. Evaluating scalable uncertainty estimation methods for deep learning-based molecular property prediction.Journal of Chemical Information and Modeling, 60(6):2697–2717, June 2020
2020
-
[57]
Ltau-ff: Loss trajectory analysis for uncertainty in atomistic force fields.Machine Learning: Science and Technology, 6(1):015048, feb 2025
Joshua A Vita, Amit Samanta, Fei Zhou, and Vincenzo Lordi. Ltau-ff: Loss trajectory analysis for uncertainty in atomistic force fields.Machine Learning: Science and Technology, 6(1):015048, feb 2025
2025
-
[58]
Fast uncertainty estimates in deep learning interatomic potentials.The Journal of Chemical Physics, 158(16), 2023
Albert Zhu, Simon Batzner, Albert Musaelian, and Boris Kozinsky. Fast uncertainty estimates in deep learning interatomic potentials.The Journal of Chemical Physics, 158(16), 2023
2023
-
[59]
Deep evidential regression.Advances in neural information processing systems, 33:14927–14937, 2020
Alexander Amini, Wilko Schwarting, Ava Soleimany, and Daniela Rus. Deep evidential regression.Advances in neural information processing systems, 33:14927–14937, 2020
2020
-
[60]
Soleimany, Alexander Amini, Samuel Goldman, Daniela Rus, Sangeeta N
Ava P. Soleimany, Alexander Amini, Samuel Goldman, Daniela Rus, Sangeeta N. Bhatia, and Connor W. Coley. Evidential deep learning for guided molecular property prediction and discovery.ACS Central Science, 7(8):1356–1367, August 2021
2021
-
[61]
Dropout as a Bayesian approximation: Representing model uncertainty in deep learning
Yarin Gal and Zoubin Ghahramani. Dropout as a Bayesian approximation: Representing model uncertainty in deep learning. InProceedings of The 33rd International Conference on Machine Learning, volume 48 of Proceedings of Machine Learning Research, pages 1050–1059. PMLR, 2016
2016
-
[62]
When gaussian process meets big data: A review of scalable gps.IEEE transactions on neural networks and learning systems, 31(11):4405–4423, 2020
Haitao Liu, Yew-Soon Ong, Xiaobo Shen, and Jianfei Cai. When gaussian process meets big data: A review of scalable gps.IEEE transactions on neural networks and learning systems, 31(11):4405–4423, 2020
2020
-
[63]
SimSon: Simple contrastive learning of SMILES for molecular property prediction.Bioinformatics, 41(5), 2025
Chae Eun Lee, Jin Sob Kim, Jin Hong Min, and Sung Won Han. SimSon: Simple contrastive learning of SMILES for molecular property prediction.Bioinformatics, 41(5), 2025
2025
-
[64]
Convolutional neural network based on SMILES representation of compounds for detecting chemical motif.BMC Bioinformatics, 19(Suppl 19):526, 2018
Masaki Hirohara, Yutaka Saito, Yuki Koda, Koji Sato, and Yasubumi Sakakibara. Convolutional neural network based on SMILES representation of compounds for detecting chemical motif.BMC Bioinformatics, 19(Suppl 19):526, 2018
2018
-
[65]
Deepsmiles: An adaptation of smiles for use in machine-learning of chemical structures.ChemRxiv, 2018(0919), 2018
Noel O’Boyle and Andrew Dalke. Deepsmiles: An adaptation of smiles for use in machine-learning of chemical structures.ChemRxiv, 2018(0919), 2018. 24
2018
-
[66]
Smiles pair encoding: A data-driven substructure tokenization algorithm for deep learning.Journal of Chemical Information and Modeling, 61(4):1560–1569, 2021
Xinhao Li and Denis Fourches. Smiles pair encoding: A data-driven substructure tokenization algorithm for deep learning.Journal of Chemical Information and Modeling, 61(4):1560–1569, 2021. PMID: 33715361
2021
-
[67]
SPVec: A Word2vec-Inspired Feature Representation Method for Drug-Target Interaction Prediction.Frontiers in Chemistry, 7, 2020
Yu-Fang Zhang, Xiangeng Wang, Aman Chandra Kaushik, Yanyi Chu, Xiaoqi Shan, Ming-Zhu Zhao, Qin Xu, and Dong-Qing Wei. SPVec: A Word2vec-Inspired Feature Representation Method for Drug-Target Interaction Prediction.Frontiers in Chemistry, 7, 2020
2020
-
[68]
AlHammadi
Obaid Khaleifah Alhmoudi, Mahmoud Aboushanab, Muhammed Thameem, Ali Elkamel, and Ali A. AlHammadi. Domain adaptation of a smiles chemical transformer to selfies with limited computational resources.Scientific Reports, 15(1):23627, 2025
2025
-
[69]
Mol-bert: An effective molecular representation with bert for molecular property prediction.Wireless Communications and Mobile Computing, 2021(1):7181815, 2021
Juncai Li and Xiaofei Jiang. Mol-bert: An effective molecular representation with bert for molecular property prediction.Wireless Communications and Mobile Computing, 2021(1):7181815, 2021
2021
-
[70]
Chemformer: a pre-trained transformer for computational chemistry.Machine Learning: Science and Technology, 3(1):015022, jan 2022
Ross Irwin, Spyridon Dimitriadis, Jiazhen He, and Esben Jannik Bjerrum. Chemformer: a pre-trained transformer for computational chemistry.Machine Learning: Science and Technology, 3(1):015022, jan 2022
2022
-
[71]
Bonggun Shin, Sungsoo Park, Keunsoo Kang, and Joyce C. Ho. Self-Attention Based Molecule Representation for Predicting Drug-Target Interaction. In Finale Doshi-Velez, Jim Fackler, Ken Jung, David Kale, Rajesh Ranganath, Byron Wallace, and Jenna Wiens, editors,Proceedings of the 4th Machine Learning for Healthcare Conference, volume 106 ofProceedings of Ma...
2019
-
[72]
Goh, Nathan O
Garrett B. Goh, Nathan O. Hodas, Charles Siegel, and Abhinav Vishnu. Smiles2vec: An interpretable general- purpose deep neural network for predicting chemical properties, 2018
2018
-
[73]
ReactionT5: a pre-trained transformer model for accurate chemical reaction prediction with limited data.Journal of Cheminformatics, 17(1):126, August 2025
Tatsuya Sagawa and Ryosuke Kojima. ReactionT5: a pre-trained transformer model for accurate chemical reaction prediction with limited data.Journal of Cheminformatics, 17(1):126, August 2025
2025
-
[74]
Chemical representation learning for toxicity prediction.Digital Discovery, 2(3):674–691, 2023
Jannis Born, Greta Markert, Nikita Janakarajan, Talia B Kimber, Andrea V olkamer, María Rodríguez Martínez, and Matteo Manica. Chemical representation learning for toxicity prediction.Digital Discovery, 2(3):674–691, 2023
2023
-
[75]
MolTrans: Molecular Interaction Transformer for drug–target interaction prediction.Bioinformatics, 37(6):830–836, March 2021
Kexin Huang, Cao Xiao, Lucas M Glass, and Jimeng Sun. MolTrans: Molecular Interaction Transformer for drug–target interaction prediction.Bioinformatics, 37(6):830–836, March 2021
2021
-
[76]
Group selfies: a robust fragment-based molecular string representation.Digital Discovery, 2, 03 2023
Austin Cheng, Andy Cai, Santiago Miret, Gustavo Malkomes, Mariano Phielipp, and Alán Aspuru-Guzik. Group selfies: a robust fragment-based molecular string representation.Digital Discovery, 2, 03 2023
2023
-
[77]
SELFormer: Molecular representation learning via SELFIES language models.Machine Learning: Science and Technology, 4(2):025035, 2023
Atakan Yüksel, Erva Ulusoy, Atabey Ünlü, and Tunca Do˘gan. SELFormer: Molecular representation learning via SELFIES language models.Machine Learning: Science and Technology, 4(2):025035, 2023
2023
-
[78]
Self-referencing embedded strings (selfies): A 100% robust molecular string representation.Machine Learning: Science and Technology, 1(4):045024, 2020
Mario Krenn, Florian Häse, AkshatKumar Nigam, Pascal Friederich, and Alan Aspuru-Guzik. Self-referencing embedded strings (selfies): A 100% robust molecular string representation.Machine Learning: Science and Technology, 1(4):045024, 2020
2020
-
[79]
Distributed representations of words and phrases and their compositionality
Tomas Mikolov, Ilya Sutskever, Kai Chen, Greg S Corrado, and Jeff Dean. Distributed representations of words and phrases and their compositionality. In C.J. Burges, L. Bottou, M. Welling, Z. Ghahramani, and K.Q. Weinberger, editors,Advances in Neural Information Processing Systems, volume 26. Curran Associates, Inc., 2013
2013
-
[80]
Heller, Alan McNaught, Igor Pletnev, Stephen Stein, and Dmitrii Tchekhovskoi
Stephen R. Heller, Alan McNaught, Igor Pletnev, Stephen Stein, and Dmitrii Tchekhovskoi. Inchi, the IUPAC international chemical identifier.Journal of Cheminformatics, 7(1):23, 2015
2015
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.