Recognition: unknown
Boltzmann Machine Learning with a Parallel, Persistent Markov chain Monte Carlo method for Estimating Evolutionary Fields and Couplings from a Protein Multiple Sequence Alignment
Pith reviewed 2026-05-10 03:22 UTC · model grok-4.3
The pith
Parallel persistent MCMC in Boltzmann machines yields reproducible estimates of evolutionary fields and couplings from protein MSAs.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Employing parallel persistent Markov chain Monte Carlo to compute marginals inside Boltzmann machine learning, together with stochastic gradient descent and conformation-based adjustment of the two regularization parameters, produces reproducible estimates of evolutionary fields and couplings from multiple sequence alignments.
What carries the argument
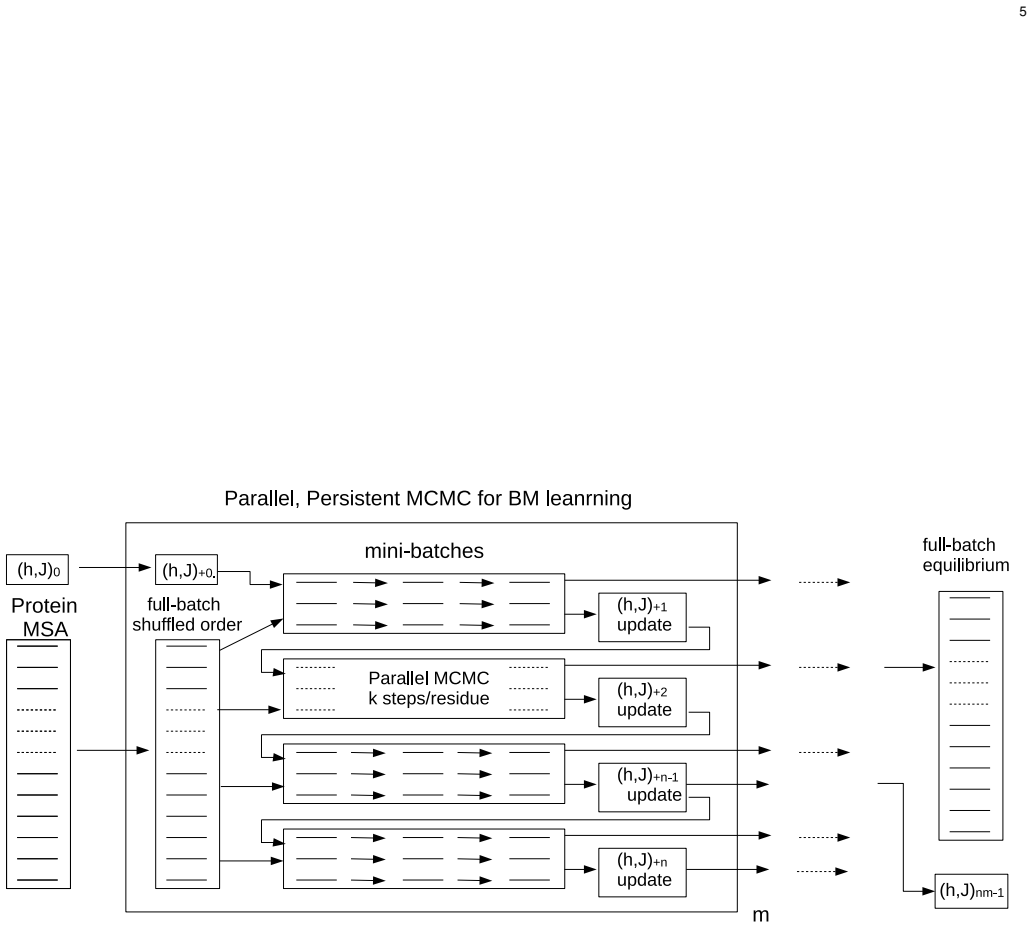
parallel persistent Markov chain Monte Carlo sampler for estimating single-site and pairwise marginal distributions during each Boltzmann machine learning step
If this is right
- Reproducible fields and couplings become available for downstream analysis of protein structure and evolution.
- The method applies directly to the eight tested protein families without depending on contact-prediction precision for tuning.
- Computational time per learning step is lowered compared with standard Boltzmann machine implementations.
- The conformation-based hyperparameter rule replaces an insensitive criterion with one tied to structural expectations.
Where Pith is reading between the lines
- The same sampler and tuning logic could be tested on larger or more divergent alignments to check scalability.
- If the conformation condition holds across more families it may serve as a default choice in other sequence-based inference tasks.
- Reproducible coupling estimates open the possibility of systematic comparison of evolutionary constraints across related proteins.
Load-bearing premise
The chosen condition for adjusting the regularization parameters on fields and couplings is the right one for generating biologically relevant values.
What would settle it
Running the full procedure independently on the same MSA multiple times and finding that the resulting fields or couplings differ by more than small numerical fluctuations would falsify the reproducibility claim.
Figures

read the original abstract
The inverse Potts problem for estimating evolutionary single-site fields and pairwise couplings in homologous protein sequences from their single-site and pairwise amino acid frequencies observed in their multiple sequence alignment would be still one of useful methods in the studies of protein structure and evolution. Since the reproducibility of fields and couplings are the most important, the Boltzmann machine method is employed here, although it is computationally intensive. In order to reduce computational time required for the Boltzmann machine, parallel, persistent Markov chain Monte Carlo method is employed to estimate the single-site and pairwise marginal distributions in each learning step. Also, stochastic gradient descent methods are used to reduce computational time for each learning. Another problem is how to adjust the values of hyperparameters; there are two regularization parameters for evolutionary fields and couplings. The precision of contact residue pair prediction is often used to adjust the hyperparameters. However, it is not sensitive to these regularization parameters. Here, they are adjusted for the fields and couplings to satisfy a specific condition that is appropriate for protein conformations. This method has been applied to eight protein families.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes using Boltzmann machine learning with a parallel, persistent Markov chain Monte Carlo (MCMC) sampler and stochastic gradient descent to estimate evolutionary fields and pairwise couplings from protein MSAs. Hyperparameters (two regularization parameters) are tuned so that the resulting fields and couplings satisfy a specific condition appropriate for protein conformations, rather than by optimizing contact-prediction precision. The approach is applied to eight protein families, with the goal of improving reproducibility over standard inverse Potts methods.
Significance. If the conformation-based hyperparameter condition can be shown to be well-defined, non-circular, and to yield biologically relevant parameters, the method could offer a computationally efficient route to reproducible evolutionary coupling estimates. The persistent MCMC component addresses a recognized bottleneck in Boltzmann machine training for Potts models and, if correctly implemented, would be a positive technical contribution.
major comments (3)
- [Abstract] Abstract: the hyperparameter adjustment is described only as tuning regularization parameters so that fields and couplings 'satisfy a specific condition that is appropriate for protein conformations.' No mathematical definition, derivation, or falsifiable test of this condition (e.g., a variance constraint, secondary-structure correlation, or moment-matching requirement) is supplied. Because this step is presented as the key alternative to contact-precision tuning, its explicit form is load-bearing for the reproducibility claim.
- [Abstract] Abstract and Methods: no equations, update rules, or pseudocode are given for the parallel persistent MCMC sampler, the stochastic gradient descent steps, or the precise regularization terms. Without these, it is impossible to verify that the implementation is correct or to reproduce the reported efficiency gains.
- [Results] Results (application to eight families): the manuscript states only that the method 'has been applied to eight protein families' but reports no quantitative metrics (contact precision, reproducibility across independent runs, comparison to baseline inverse Potts or other BM implementations, or error bars). This absence prevents evaluation of whether the conformation condition actually produces biologically relevant fields and couplings.
minor comments (1)
- [Abstract] The abstract is overly condensed; separating the description of the MCMC sampler, the hyperparameter rule, and the empirical application into distinct sentences would improve readability.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which have helped us identify areas where the manuscript can be improved for clarity and completeness. We provide point-by-point responses to the major comments and outline the revisions we will make.
read point-by-point responses
-
Referee: [Abstract] Abstract: the hyperparameter adjustment is described only as tuning regularization parameters so that fields and couplings 'satisfy a specific condition that is appropriate for protein conformations.' No mathematical definition, derivation, or falsifiable test of this condition (e.g., a variance constraint, secondary-structure correlation, or moment-matching requirement) is supplied. Because this step is presented as the key alternative to contact-precision tuning, its explicit form is load-bearing for the reproducibility claim.
Authors: We agree that the abstract does not provide the mathematical definition of the condition. We will revise the abstract to include the explicit form of the condition used for tuning the regularization parameters, along with its derivation and a description of the falsifiable test in the Methods section. This will clarify its role as an alternative to contact-precision tuning and address concerns about its definition and non-circularity. revision: yes
-
Referee: [Abstract] Abstract and Methods: no equations, update rules, or pseudocode are given for the parallel persistent MCMC sampler, the stochastic gradient descent steps, or the precise regularization terms. Without these, it is impossible to verify that the implementation is correct or to reproduce the reported efficiency gains.
Authors: We acknowledge this omission in the submitted version. The revised manuscript will include the mathematical formulation of the parallel persistent MCMC sampler, including the update rules for maintaining persistent chains and the stochastic gradient descent procedure for parameter updates. We will also provide the explicit forms of the regularization terms and pseudocode for the overall algorithm in the Methods section to facilitate verification and reproduction of the efficiency gains. revision: yes
-
Referee: [Results] Results (application to eight families): the manuscript states only that the method 'has been applied to eight protein families' but reports no quantitative metrics (contact precision, reproducibility across independent runs, comparison to baseline inverse Potts or other BM implementations, or error bars). This absence prevents evaluation of whether the conformation condition actually produces biologically relevant fields and couplings.
Authors: The initial manuscript presents the application to eight families primarily to illustrate the method's feasibility. We agree that quantitative metrics are necessary to fully evaluate the approach. In the revised Results section, we will report contact prediction precisions, reproducibility measures across multiple independent runs, comparisons to standard inverse Potts methods and other Boltzmann machine implementations, and associated error bars for the eight protein families. This will enable a direct assessment of the biological relevance of the inferred parameters under the conformation-based condition. revision: yes
Circularity Check
No significant circularity detected in the derivation chain
full rationale
The paper presents a computational procedure: parallel persistent MCMC within Boltzmann machine learning, combined with stochastic gradient descent, to estimate fields and couplings from observed frequencies in an MSA. The hyperparameter adjustment step is described as tuning regularization parameters so that the resulting fields and couplings 'satisfy a specific condition that is appropriate for protein conformations,' after rejecting contact-precision tuning on grounds of insensitivity. No equation, functional form, or derivation is supplied in the given text that defines this condition in terms of the model outputs themselves or that reduces the final estimates to the inputs by construction. The core estimation steps rely on standard MCMC sampling and gradient updates whose correctness is independent of the target result. No self-citation load-bearing step, fitted-input-as-prediction, or ansatz smuggling is exhibited. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- regularization parameters for fields and couplings
Reference graph
Works this paper leans on
-
[1]
Using sequence alignments to predict protein structure and stability with high accuracy,
A. Lapedes, B. Giraud, and C. Jarzynsk, “Using sequence alignments to predict protein structure and stability with high accuracy,”LANL Sciece Magazine, vol. LA-UR-02-4481, 2002
2002
-
[2]
Using sequence alignments to predict protein structure and stability with high accuracy,
——, “Using sequence alignments to predict protein structure and stability with high accuracy,”arXiv:1207.2484 [q-bio.QM], 2012
-
[3]
Direct-coupling analysis of residue coevolution captures native contacts across many protein families,
F. Morcos, A. Pagnani, B. Lunt, A. Bertolino, D. S. Marks, C. Sander, R. Zecchina, J. N. Onuchic, T. Hwa, and M. Weigt, “Direct-coupling analysis of residue coevolution captures native contacts across many protein families,”Proc. Natl. Acad. Sci. USA, vol. 108, pp. E1293–E1301, 2011
2011
-
[4]
Protein 3D structure computed from evolutionary sequence variation,
D. S. Marks, L. J. Colwell, R. Sheridan, T. A. Hopf, A. Pagnani, R. Zecchina, and C. Sander, “Protein 3D structure computed from evolutionary sequence variation,”PLoS ONE, vol. 6, no. 12, p. e28766, 12 2011. [Online]. Available: http://dx.doi.org/10.1371/journal.pone.0028766
-
[5]
Improved contact prediction in proteins: Using pseudolikelihoods to infer Potts models,
M. Ekeberg, C. L ¨ovkvist, Y . Lan, M. Weigt, and E. Aurell, “Improved contact prediction in proteins: Using pseudolikelihoods to infer Potts models,” Phys. Rev. E, vol. 87, p. 012707, 2013. [Online]. Available: http://link.aps.org/doi/10.1103/PhysRevE.87.012707
-
[6]
Fast pseudolikelihood maximization for direct-coupling analysis of protein structure from many homologous amino-acid sequences,
M. Ekeberg, T. Hartonen, and E. Aurell, “Fast pseudolikelihood maximization for direct-coupling analysis of protein structure from many homologous amino-acid sequences,”J. Comput. Phys., vol. 276, pp. 341–356, 2014
2014
-
[7]
Critical assessment of methods of protein structure prediction: Progress and new directions in round XI,
J. Moult, K. Fidelis, A. Kryshtafovych, T. Schwede, and A. Tramontano, “Critical assessment of methods of protein structure prediction: Progress and new directions in round XI,”Proteins, vol. 84(S1), pp. 4–14, 2016
2016
-
[8]
ACE: adaptive cluster expansion for maximum entropy graphical model inference,
J. P. Barton, E. D. Leonardis, A. Coucke, and S. Cocco, “ACE: adaptive cluster expansion for maximum entropy graphical model inference,”Bioinformatics, vol. 32, pp. 3089–3097, 2016
2016
-
[9]
Inverse statistical physics of protein sequences: A key issues review,
S. Cocco, C. Feinauer, M. Figliuzzi, R. Monasson, and M. Weigt, “Inverse statistical physics of protein sequences: A key issues review,”arXiv:1703.01222 [q-bio.BM], 2017
-
[10]
How pairwise coevolutionary models capture the collective residue variability in proteins?
M. Figliuzzi, P. Barrat-Charlaix, and M. Weigt, “How pairwise coevolutionary models capture the collective residue variability in proteins?”Mol. Biol. Evol., vol. 35, pp. 1018–1027, 2018
2018
-
[11]
Optimal perceptual inference,
G. E. Hinton and T. J. Sejnowski, “Optimal perceptual inference,”Proceedings of the IEEE conference on Computer Vision and Pattern Recognition, pp. 448–453, 1983
1983
-
[12]
A learning algorithm for Boltzmann machines,
A. D. H., H. G. E., and S. T. J., “A learning algorithm for Boltzmann machines,”Cogn. Sci., vol. 9, pp. 147–169, 1985
1985
-
[13]
Boltzmann machine,
G. E. Hinton, “Boltzmann machine,”Scholarpedia, vol. 2, p. 1668, 2007
2007
-
[14]
Identification of direct residue contacts in protein-protein interaction by message passing,
M. Weigt, R. A. White, H. Szurmant, J. A. Hoch, and T. Hwa, “Identification of direct residue contacts in protein-protein interaction by message passing,” Proc. Natl. Acad. Sci. USA, vol. 106, pp. 67–72, 2009
2009
-
[15]
Selection originating from protein stability/foldability: Relationships between protein folding free energy, sequence ensemble, and fitness,
S. Miyazawa, “Selection originating from protein stability/foldability: Relationships between protein folding free energy, sequence ensemble, and fitness,” J. Theor . Biol., vol. 433, pp. 21–38, 2017
2017
-
[16]
——, “Boltzmann machine learning and regularization methods for inferring evolutionary fields and couplings from a multiple sequence alignment,” arXiv:1909.05006, 2019
-
[17]
Boltzmann machine learning and regularization methods for inferring evolutionary fields and couplings from a multiple sequence alignment,
——, “Boltzmann machine learning and regularization methods for inferring evolutionary fields and couplings from a multiple sequence alignment,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 19, pp. 328–342, 2020
2020
-
[18]
Equation of state calculations by fast computing machines
N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller, “Equation of state calculations by fast computing machines.”J. Chem. Phys., vol. 21, pp. 1087–1092, 1953
1953
-
[19]
Monte Carlo sampling methods using Markov chains and their applications,
W. K. Hastings, “Monte Carlo sampling methods using Markov chains and their applications,”Biometrika, vol. 57, pp. 97–109, 1970
1970
-
[20]
Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images,
S. Geman and D. Geman, “Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images,”IEEE Trans. Pattern Anal. Mach. Intell., 1984
1984
-
[21]
Training products of experts by minimizing contrastive divergence,
G. E. Hinton, “Training products of experts by minimizing contrastive divergence,”Neural Computation, vol. 14, pp. 1771–1800, 2002
2002
-
[22]
A fast learning algorithm for deep belief nets,
G. E. Hinton, S. Osindero, and Y .-W. Teh, “A fast learning algorithm for deep belief nets,”Neural Computation, vol. 18, pp. 1527–1554, 2006
2006
-
[23]
Justifying and generalizing contrastive divergence,
Y . Bengio and O. Delalleau, “Justifying and generalizing contrastive divergence,”Neural Computation, vol. 21, pp. 1601–1621, 2009
2009
-
[24]
Training restricted Boltzmann machines using approximations to the likelihood gradient,
T. Tieleman, “Training restricted Boltzmann machines using approximations to the likelihood gradient,” inICML ’08: Proceedings of the 25th international conference on Machine learning, 2008, pp. 1064–1071
2008
-
[25]
Prediction of contact residue pairs based on co-substitution between sites in protein structures,
S. Miyazawa, “Prediction of contact residue pairs based on co-substitution between sites in protein structures,”PLoS ONE, vol. 8, no. 1, p. e54252, 01
-
[26]
Available: http://dx.doi.org/10.1371/journal.pone.0054252
[Online]. Available: http://dx.doi.org/10.1371/journal.pone.0054252
-
[27]
Prediction of structures and interactions from genome information,
——, “Prediction of structures and interactions from genome information,”arXiv:1709.08021 [q-bio.BM], 2017
-
[28]
Prediction of structures and interactions from genome information,
——, “Prediction of structures and interactions from genome information,” inIntegrative Structural Biology with Hybrid Methods, ser. Advances in Experimental Medicine and Biology 1105, H. Nakamura, Ed. Singapore: Springer Nature Singapore Pte Ltd., 2018, ch. 9
2018
-
[29]
C. Baldassi, M. Zamparo, C. Feinauer, A. Procaccini, R. Zecchina, M. Weigt, and A. Pagnani, “Fast and accurate multivariate Gaussian modeling of protein families: Predicting residue contacts and protein-interaction partners,”PLoS ONE, vol. 9, no. 3, p. e92721, 03 2014. [Online]. Available: https://dx.doi.org/10.1371/journal.pone.0092721
-
[30]
Learning generative models for protein fold families,
S. Balakrishnan, H. Kamisetty, J. G. Carbonell, S. I. Lee, and C. J. Langmead, “Learning generative models for protein fold families,”Proteins, vol. 79, pp. 1061–1078, 2011
2011
-
[31]
Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era,
H. Kamisetty, S. Ovchinnikov, and D. Baker, “Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era,”Proc. Natl. Acad. Sci. USA, vol. 110, pp. 15 674–15 679, 2013
2013
-
[32]
Learning protein constitutive motifs from sequence data,
J. Tubiana, S. Cocco, and R. Monasson, “Learning protein constitutive motifs from sequence data,”eLife, vol. 8, p. e39397, 2019
2019
-
[33]
Selection of sequence motifs and generative Hopfield-Potts models for protein families,
K. Shimagaki and M. Weigt, “Selection of sequence motifs and generative Hopfield-Potts models for protein families,”Phys. Rev. E, vol. 100, p. 032128, 2019
2019
-
[34]
Equilibrium folding and unfolding pathways for a model protein,
S. Miyazawa and R. L. Jernigan, “Equilibrium folding and unfolding pathways for a model protein,”Biopolymers, vol. 21, pp. 1333–1363, 1982
1982
-
[35]
M. Schmidt. (2017) CPSC 540: Machine learning; group L1-regularization, proximal-gradient. [Online]. Available: https://www.cs.ubc.ca/ ∼schmidtm/ Courses/540-W17/L5.pdf
2017
-
[36]
Wilkinson
D. Wilkinson. (2019) Discussion of: Unbiased MCMC with couplings (Jacob et al). [Online]. Available: https://darrenjw.wordpress.com/2019/12/12/ unbiased-mcmc-with-couplings/
2019
-
[37]
Exact estimation for Markov chain equilibrium expectations,
P. W. Glynn and C.-h. Rhee, “Exact estimation for Markov chain equilibrium expectations,”Journal of Applied Probability, vol. 51(A), pp. 377–389, 2014
2014
-
[38]
Adam: A Method for Stochastic Optimization
D. P. Kingma and J. L. Ba, “Adam: A method for stochastic optimization,”arXiv:1412.6980, 2014
work page internal anchor Pith review Pith/arXiv arXiv 2014
-
[39]
M. Schmidt. (2017) CPSC 540: Machine learning; structured sparsity, stochastic subgradient. [Online]. Available: https://www.cs.ubc.ca/ ∼schmidtm/ Courses/540-W17/L6.pdf
2017
-
[40]
A new approach to the design of stable proteins,
E. I. Shakhnovich and A. M. Gutin, “A new approach to the design of stable proteins,”Protein Eng., vol. 6, pp. 793–800, 1993
1993
-
[41]
Engineering of stable and fast-folding sequences of model proteins,
——, “Engineering of stable and fast-folding sequences of model proteins,”Proc. Natl. Acad. Sci. USA, vol. 90, pp. 7195–7199, 1993
1993
-
[42]
Statistical mechanics of proteins with evolutionary selected sequences,
S. Ramanathan and E. Shakhnovich, “Statistical mechanics of proteins with evolutionary selected sequences,”Phys. Rev. E, vol. 50, pp. 1303–1312, 1994
1994
-
[43]
Statistical mechanics of simple models of protein folding and design,
V . S. Pande, A. Y . Grosberg, and T. Tanaka, “Statistical mechanics of simple models of protein folding and design,”Biophys. J., vol. 73, pp. 3192–3210, 1997. Sanzo Miyazawahad worked for the Graduate School of Engineering in Gunma University, Japan until retired at age 65 in 2013. His research interests include protein structure and evolution. S-1 Suppl...
1997
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.