Recognition: unknown
Integrating opportunities and parametrized signatures for improved mutational processes estimation in extended sequence contexts
Pith reviewed 2026-05-08 13:14 UTC · model grok-4.3
The pith
Combining mutational opportunities, extended contexts, negative binomial modeling and parametrized signatures produces robust mutational signatures.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
We show that the combination of these four extensions gives very robust and reliable mutational signatures. In particular, we highlight the importance of including mutational opportunities and parametrizing the signatures when the mutation types describe an extended sequence context with two or three flanking nucleotides to each side of the base substitution.
What carries the argument
The parametrized signatures that incorporate mutational opportunities within an extended sequence context modeled by the negative binomial distribution.
If this is right
- Signatures estimated from extended contexts become more stable once opportunities adjust for local sequence composition.
- Parametrization lowers the effective number of free parameters, reducing overfitting as context length increases.
- The negative binomial likelihood handles overdispersion in count data more accurately than a Poisson assumption.
- The integrated approach yields signatures that more closely reflect true mutational processes rather than sampling or compositional artifacts.
Where Pith is reading between the lines
- The method could be applied to large cancer cohorts to identify subtle mutational processes that standard tools miss in noisy long-context data.
- A direct simulation study with known input signatures would quantify the exact gain in accuracy over baseline approaches.
- Wider use in genomics pipelines could sharpen attribution of mutations to specific exposures such as UV damage or chemotherapy.
Load-bearing premise
That incorporating mutational opportunities and parametrizing signatures will improve reliability without introducing bias or overfitting when the sequence context is extended to two or three flanking nucleotides, and that the negative binomial distribution adequately captures variation in the mutation counts.
What would settle it
Generate synthetic mutation counts from known ground-truth signatures in extended contexts, then compare recovery error of the four-extension method against the standard method that omits opportunities and parametrization.
Figures



read the original abstract
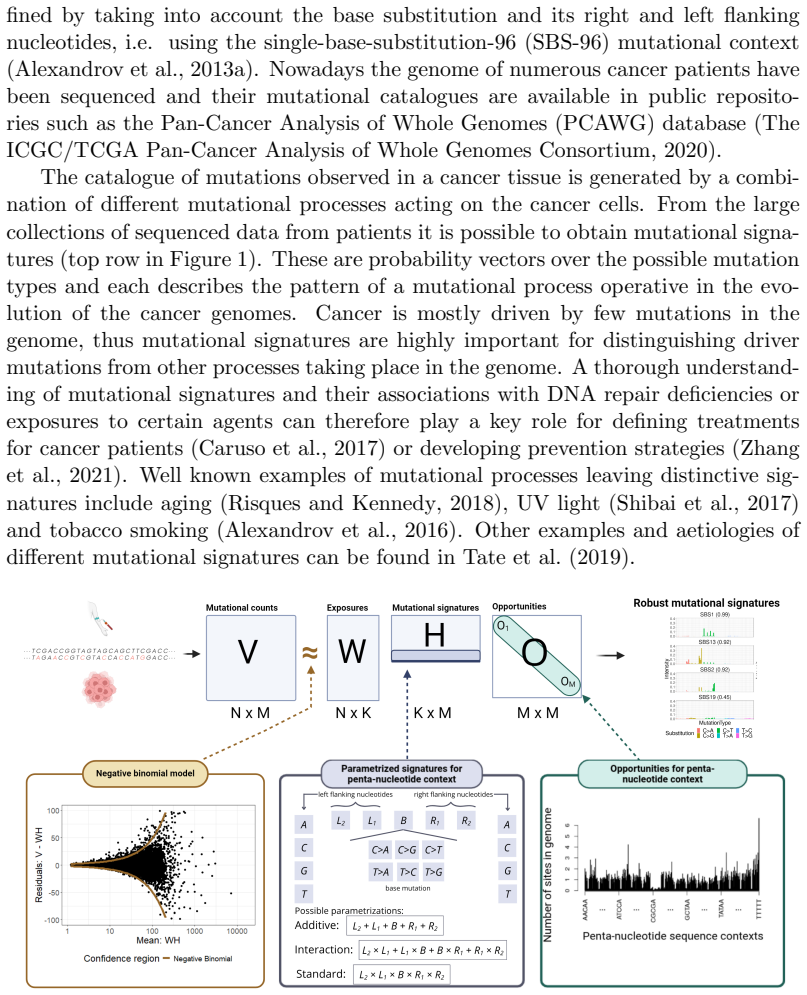
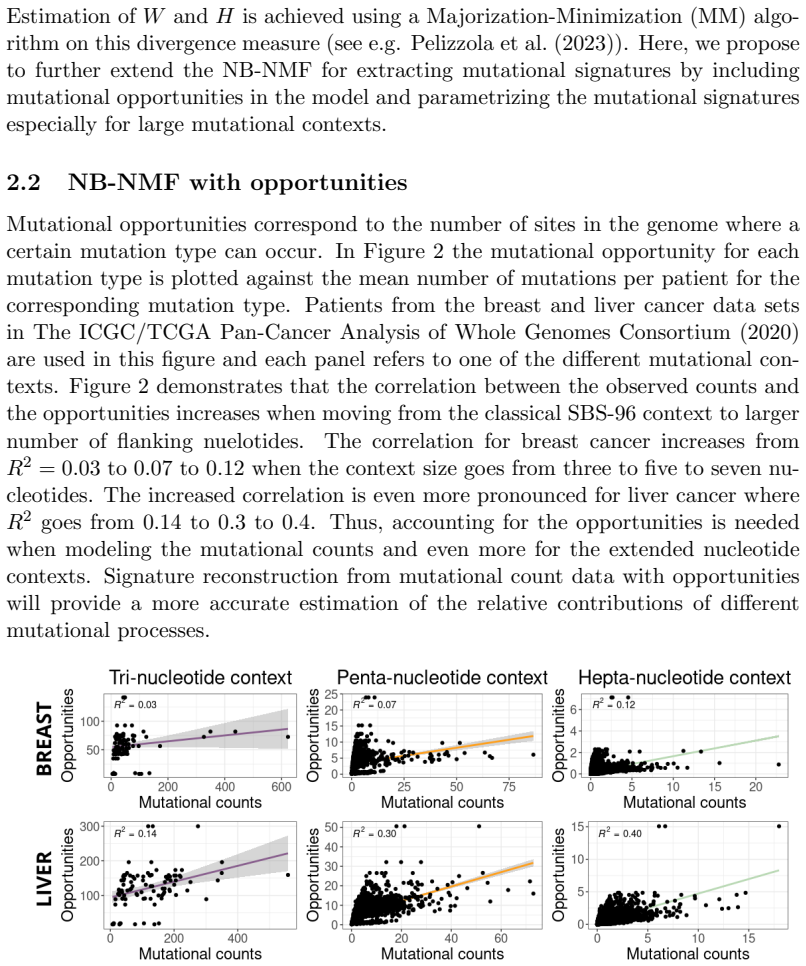
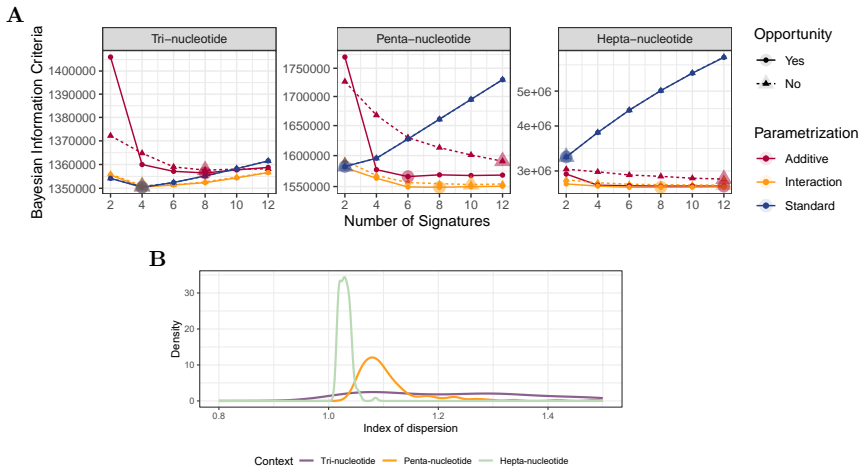
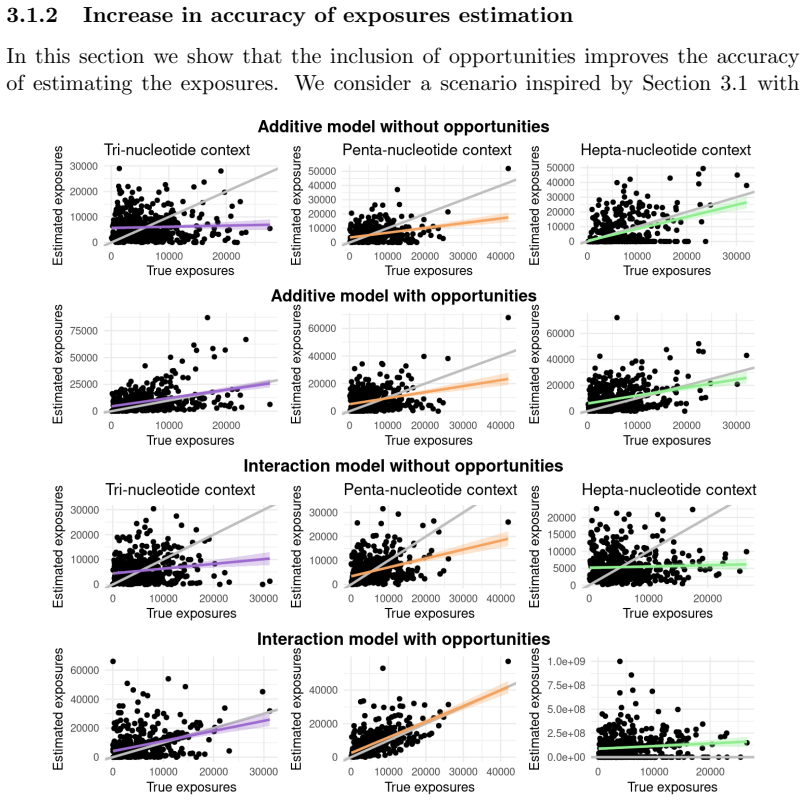
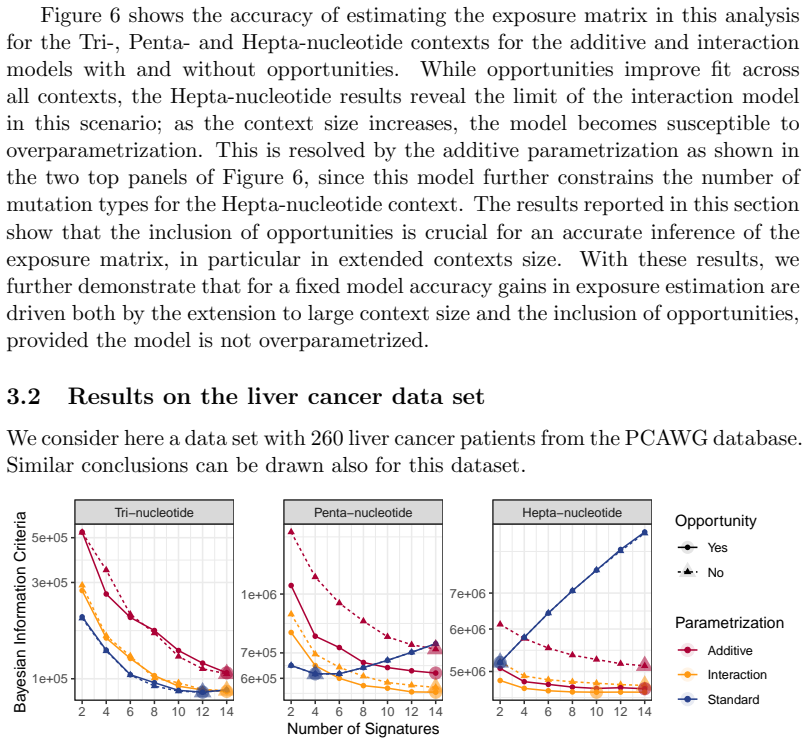
Mutational signatures describe the pattern of mutations over the different mutation types. Each mutation type is determined by a base substitution and the flanking nucleotides to the left and right of that base substitution. Due to the widespread interest in mutational signatures, several efforts have been devoted to the development of methods for robust and stable signature estimation. Here, we combine various extensions of the standard framework to estimate mutational signatures. These extensions include (a) incorporating opportunities to the analysis, (b) allowing for extended sequence contexts, (c) using the Negative Binomial model, and (d) parametrizing the signatures. We show that the combination of these four extensions gives very robust and reliable mutational signatures. In particular, we highlight the importance of including mutational opportunities and parametrizing the signatures when the mutation types describe an extended sequence context with two or three flanking nucleotides to each side of the base substitution.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes combining four extensions to standard mutational signature estimation—incorporating mutational opportunities, allowing extended sequence contexts (5-mers and 7-mers), replacing the Poisson with a Negative Binomial model, and parametrizing the signature profiles themselves—and claims that this integrated framework yields substantially more robust and reliable signatures than conventional approaches, with opportunities and parametrization being especially critical for higher-order contexts.
Significance. If the reported robustness holds, the work would provide a practical methodological improvement for extracting mutational processes from sparse count data in high-dimensional contexts, a common bottleneck in cancer genomics. The manuscript supplies simulation studies, cross-cohort stability analyses, and direct parametrized vs. non-parametrized comparisons that demonstrate variance reduction without detectable bias inflation, together with explicit dispersion estimation and goodness-of-fit diagnostics justifying the Negative Binomial; these elements strengthen the case for adoption provided the findings generalize to independent datasets.
minor comments (3)
- The abstract states that the four extensions together produce 'very robust and reliable' signatures but does not include any quantitative summary statistics (e.g., average cosine similarity, variance reduction factors, or reconstruction error) that appear in the results; adding one or two such numbers would make the headline claim immediately verifiable.
- Notation for the parametrized signatures (e.g., how the free parameters are defined for 5-mer and 7-mer contexts) is introduced without an explicit small example table showing the mapping from mutation type to parameter; a single illustrative table would improve readability.
- The manuscript compares the integrated model to the standard framework but does not report a head-to-head benchmark against other recently published extended-context methods (e.g., those using hierarchical Dirichlet processes or tensor decompositions); a brief discussion of relative performance would help situate the contribution.
Simulated Author's Rebuttal
We thank the referee for the positive and accurate summary of our manuscript, the recognition of its potential significance for mutational signature estimation in high-dimensional contexts, and the recommendation for minor revision. We address the report below.
Circularity Check
No significant circularity; extensions validated independently
full rationale
The paper extends the standard mutational signature model by four components (opportunities, extended 5/7-mer contexts, Negative Binomial likelihood, and parametric signature forms). The headline claim of improved robustness is supported by explicit simulation studies that generate data under known ground-truth signatures, cross-cohort stability comparisons, and direct side-by-side evaluation of parametrized versus non-parametrized fits that quantify variance reduction without bias inflation. The Negative Binomial is justified by estimated dispersion parameters and goodness-of-fit diagnostics on the observed counts. No equation or result is shown to equal its own fitted inputs by construction, and no load-bearing premise reduces to a self-citation chain or an unverified ansatz. The derivation therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- signature parameters
axioms (1)
- domain assumption Negative Binomial distribution is appropriate for modeling mutation counts
Reference graph
Works this paper leans on
-
[1]
B., Ju, Y
Alexandrov, L. B., Ju, Y. S., Haase, K., Van Loo, P., Martincorena, I., Nik-Zainal, S., Totoki, Y., Fujimoto, A., Nakagawa, H., Shibata, T., Campbell, P. J., Vineis, P., Phillips, D. H., and Stratton, M. R. (2016). Mutational signatures associated with tobacco smoking in human cancer.Science, 354(6312):618–622
2016
-
[2]
Stratton, M. R. (2020). The repertoire of mutational signatures in human cancer. Nature, 578(7793):94–101
2020
-
[3]
and Gori, K
Baez-Ortega, A. and Gori, K. (2017). Computational approaches for discovery of mutational signatures in cancer.Briefings in Bioinformatics, 20(1):77–88
2017
-
[4]
Bethune, J., Kleppe, A., and Besenbacher, S. (2022). A method to build extended sequence context models of point mutations and indels.Nature Communications, 13(1)
2022
-
[5]
Smith, E. S. J., Gerstung, M., Campbell, P. J., Murchison, E. P., Stratton, M. R., and Martincorena, I. (2022). Somatic mutation rates scale with lifespan across mammals.Nature, 604(7906):517–524
2022
-
[6]
B., and Tomao, F
Caruso, D., Papa, A., Tomao, S., Vici, P., Panici, P. B., and Tomao, F. (2017). Niraparib in ovarian cancer: results to date and clinical potential.Therapeutic Advances in Medical Oncology, 9(9):579–588
2017
-
[7]
S., Allen, E
Lander, E. S., Allen, E. M. V., and Sunyaev, S. R. (2020). Identification of cancer driver genes based on nucleotide context.Nature Genetics, 52(2):208–218
2020
-
[8]
J., Campbell, P
Fischer, A., Illingworth, C. J., Campbell, P. J., and Mustonen, V. (2013). EMu: Probabilistic inference of mutational processes and their localization in the cancer genome.Genome Biology, 14(4):1–10. 21
2013
-
[9]
Gori, K. and Baez-Ortega, A. (2020). sigfit: flexible bayesian inference of mutational signatures.bioRxiv, https://doi.org/10.1101/372896
-
[10]
Gouvert, O., Oberlin, T., and Fevotte, C. (2020). Negative Binomial Matrix Fac- torization.IEEE Signal Processing Letters, 27:815–819
2020
-
[11]
Lal, A., Liu, K., Tibshirani, R., Sidow, A., and Ramazzotti, D. (2021). De novo mutational signature discovery in tumor genomes using SparseSignatures.PLOS Computational Biology, 17(6):e1009119
2021
-
[12]
Laursen, R., Maretty, L., and Hobolth, A. (2024). Flexible model-based non-negative matrix factorization with application to mutational signatures.Statistical Appli- cations in Genetics and Molecular Biology, 23(1):20230034
2024
-
[13]
Lee, D. D. and Seung, H. S. (1999). Learning the parts of objects by non-negative matrix factorization.Nature, 401(6755):788–791
1999
-
[14]
Lindberg, M., Bostr¨ om, M., Elliott, K., and Larsson, E. (2019). Intragenomic vari- ability and extended sequence patterns in the mutational signature of ultraviolet light.Proceedings of the National Academy of Sciences, 116(41):20411–20417
2019
-
[15]
Lochovsky, L., Zhang, J., Fu, Y., Khurana, E., and Gerstein, M. (2015). LARVA: an integrative framework for large-scale analysis of recurrent variants in noncoding annotations.Nucleic acids research, 43(17):8123–8134
2015
-
[16]
Lyu, X., Garret, J., R¨ atsch, G., and Lehmann, K. V. (2020). Mutational signa- ture learning with supervised negative binomial non-negative matrix factorization. Bioinformatics, 36(Suppl 1):i154–i160
2020
-
[17]
Omichessan, H., Severi, G., and Perduca, V. (2019). Computational tools to detect signatures of mutational processes in DNA from tumours: A review and empirical comparison of performance.PLOS ONE, 14(9):e0221235
2019
-
[18]
Pelizzola, M., Laursen, R., and Hobolth, A. (2023). Model selection and robust inference of mutational signatures using negative binomial non-negative matrix factorization.BMC Bioinformatics, 24(1)
2023
-
[19]
A., Sørensen, S
Poulsgaard, G. A., Sørensen, S. G., Juul, R. I., Nielsen, M. M., and Pedersen, J. S. (2023). Sequence dependencies and mutation rates of localized mutational processes in cancer.Genome Medicine, 15(1)
2023
-
[20]
Risques, R. A. and Kennedy, S. R. (2018). Aging and the rise of somatic cancer- associated mutations in normal tissues.PLOS Genetics, 14(1)
2018
-
[21]
Shibai, A., Takahashi, Y., Ishizawa, Y., Motooka, D., Nakamura, S., Ying, B.-W., and Tsuru, S. (2017). Mutation accumulation under UV radiation in Escherichia coli.Scientific Reports, 7(1):1–12
2017
-
[22]
Shiraishi, Y., Tremmel, G., Miyano, S., and Stephens, M. (2015). A simple model- based approach to inferring and visualizing cancer mutation signatures.PLOS Genetics, 11(12):e1005657. 22
2015
-
[23]
Inouye, M. (1966). Frameshift mutations and the genetic code.Cold Spring Harbor Symposia on Quantitative Biology, 31(0):77–84
1966
-
[24]
E., Stefancsik, R., Thompson, S
Speedy, H. E., Stefancsik, R., Thompson, S. L., Wang, S., Ward, S., Campbell, P. J., and Forbes, S. A. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer.Nucleic Acids Research, 47(D1):D941–D947. The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020). Pan-cancer analysis of whole genomes.Nature, 578(7793):82–93. V¨ ohringer, H., Ho...
2019
-
[25]
Weinhold, N., Jacobsen, A., Schultz, N., Sander, C., and Lee, W. (2014). Genome- wide analysis of noncoding regulatory mutations in cancer.Nature genetics, 46(11):1160–1165
2014
-
[26]
S., Carter, H., Ried, T., Kim, C
Pommier, Y., Lan, Q., Rothman, N., Almeida, J. S., Carter, H., Ried, T., Kim, C. F., Lopez-Bigas, N., Garcia-Closas, M., Shi, J., Boss´ e, Y., Zhu, B., Gordenin, D. A., Alexandrov, L. B., Chanock, S. J., Wedge, D. C., and Landi, M. T. (2021). Genomic and evolutionary classification of lung cancer in never smokers.Nature Genetics, 53(9):1348–1359. 23
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.