Recognition: unknown
Tail-Greedy Unbalanced Haar Wavelet Segmentation for Copy Number Alteration Data
Pith reviewed 2026-05-08 09:20 UTC · model grok-4.3
The pith
A dual-thresholding strategy in tail-greedy unbalanced Haar wavelets improves detection of short copy number alterations in noisy sequencing data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
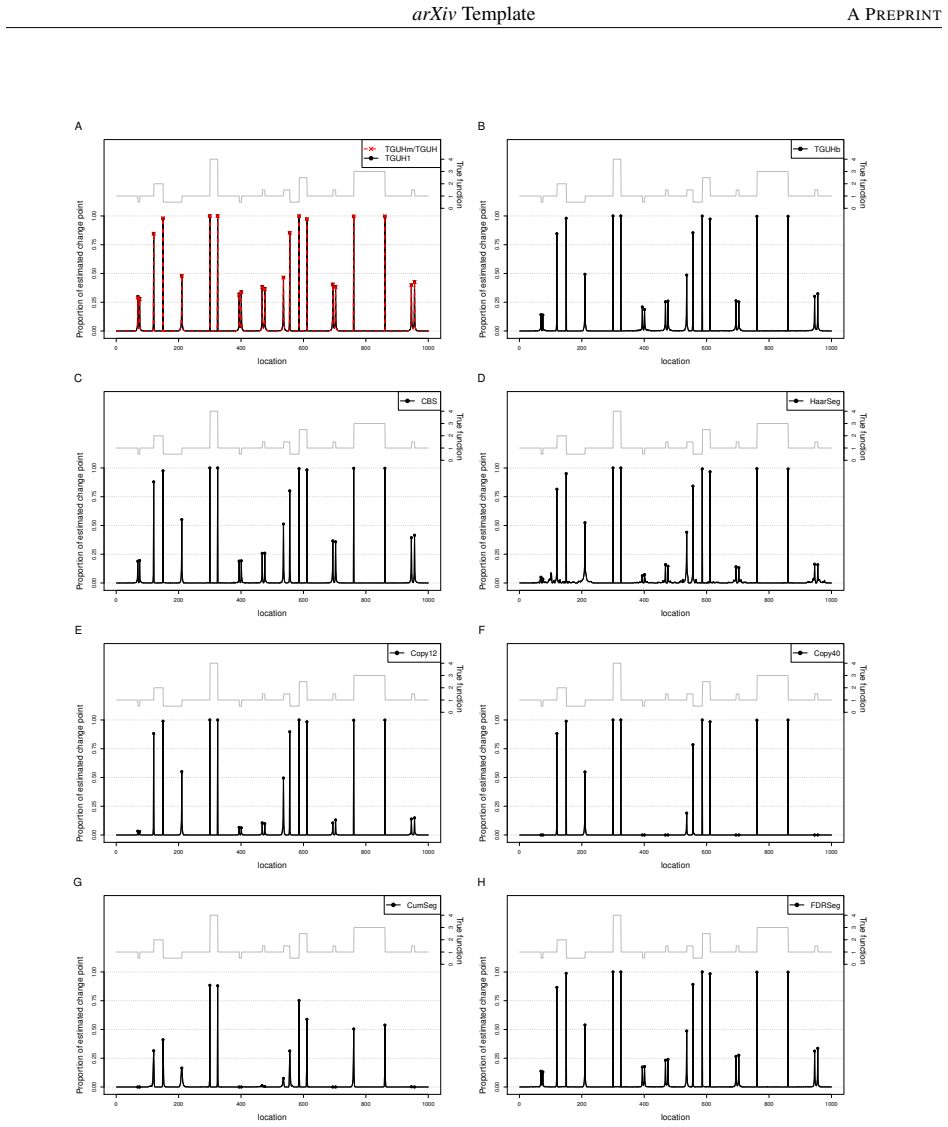
The central claim is that the TGUHm method with dual-thresholding effectively suppresses spurious spikes in the segmentation process while preserving sensitivity to short and long CNA segments, resulting in superior performance in detecting copy number alterations compared to existing methods.
What carries the argument
The tail-greedy unbalanced Haar wavelet segmentation enhanced with a dual-thresholding strategy, which applies two levels of thresholds to control the detection of segment boundaries and suppress noise-induced artifacts.
If this is right
- Improved accuracy in identifying short CNA segments under various noise conditions.
- Lower false positive rates in segmentation of noisy next-generation sequencing data.
- More reliable identification of cancer-related genes from genomic profiles.
- Competitive performance across both simulated and real datasets.
Where Pith is reading between the lines
- The dual-thresholding could generalize to other wavelet-based segmentation tasks beyond CNA detection.
- Future work might test the method on datasets with different sequencing depths or error profiles.
- Integration with machine learning could further enhance segment boundary detection.
Load-bearing premise
The noise patterns in new datasets will resemble the Gaussian, heavy-tailed, or single real cancer dataset used in the evaluations, allowing the dual thresholds to balance sensitivity and specificity effectively.
What would settle it
Apply the method to a new cancer dataset with known short CNAs and higher or different noise levels; if short segments are missed or false positives increase significantly beyond the reported rates, the improvement claim would not hold.
Figures

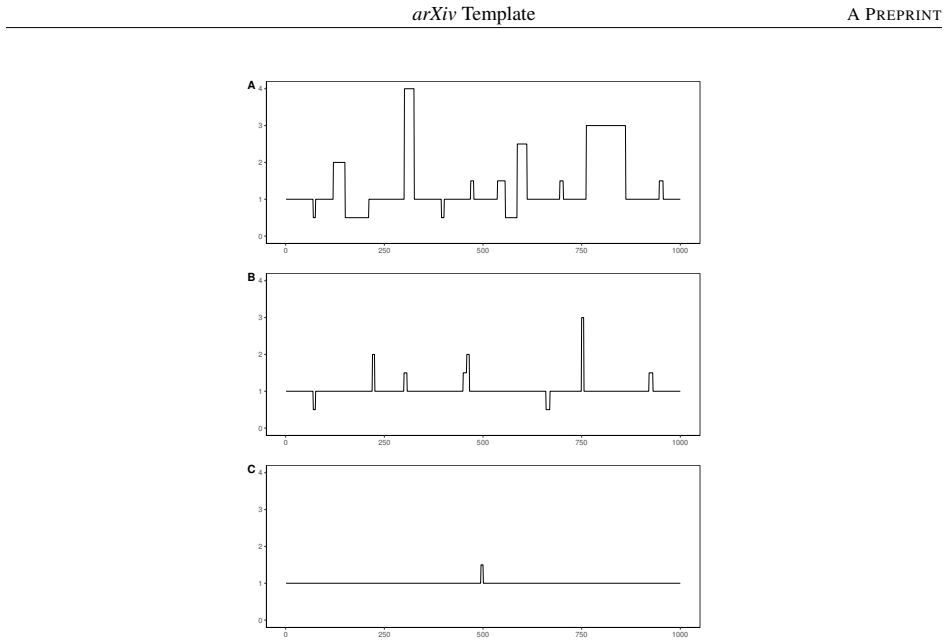
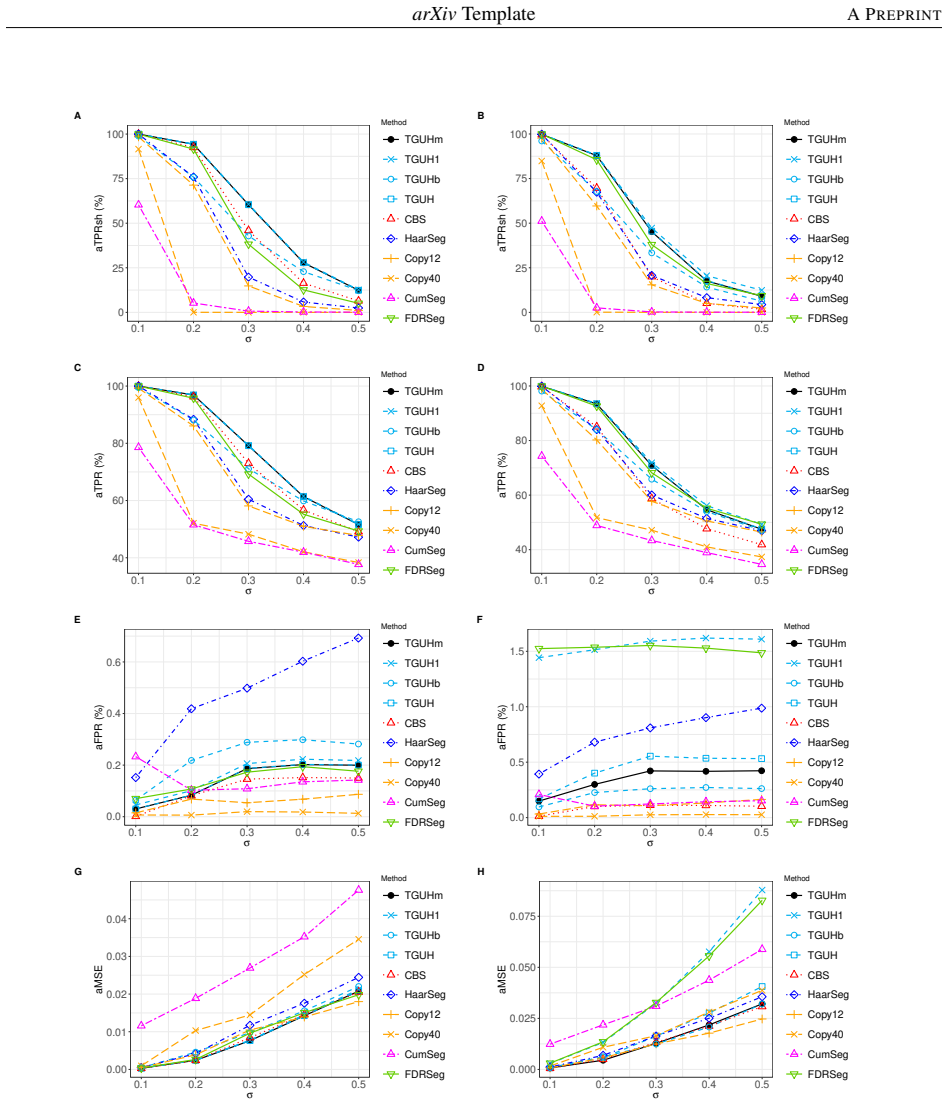
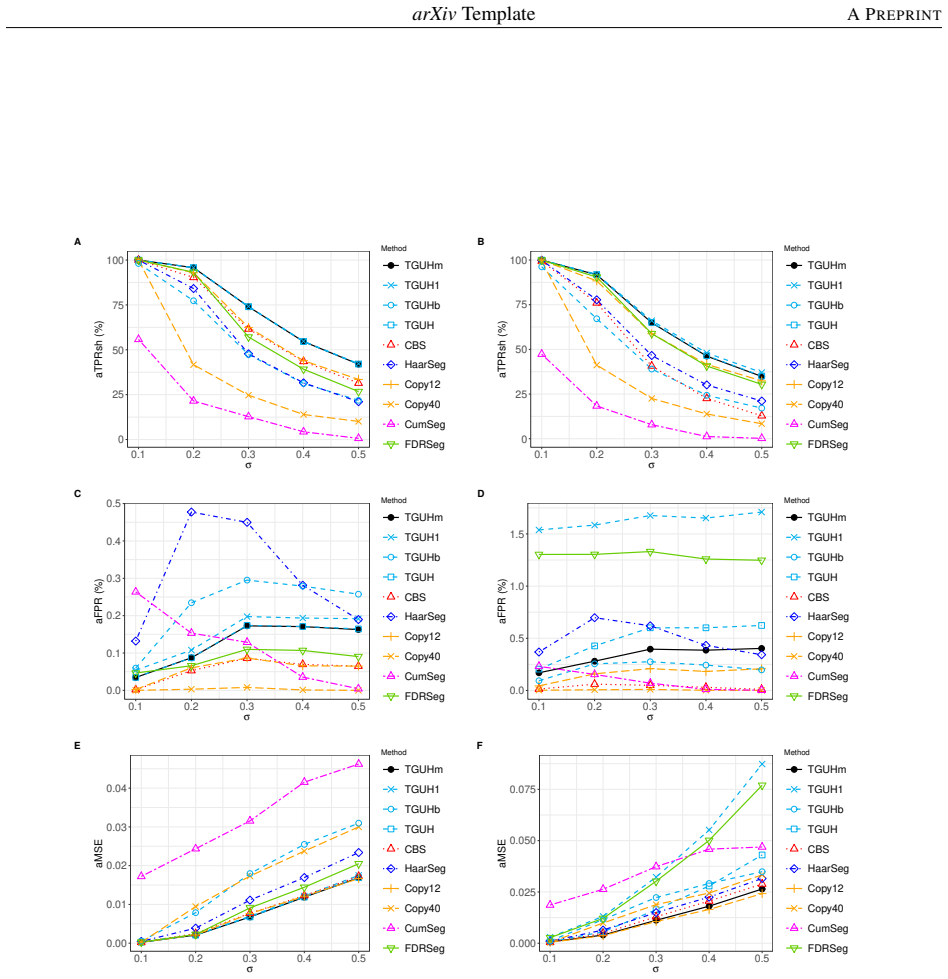

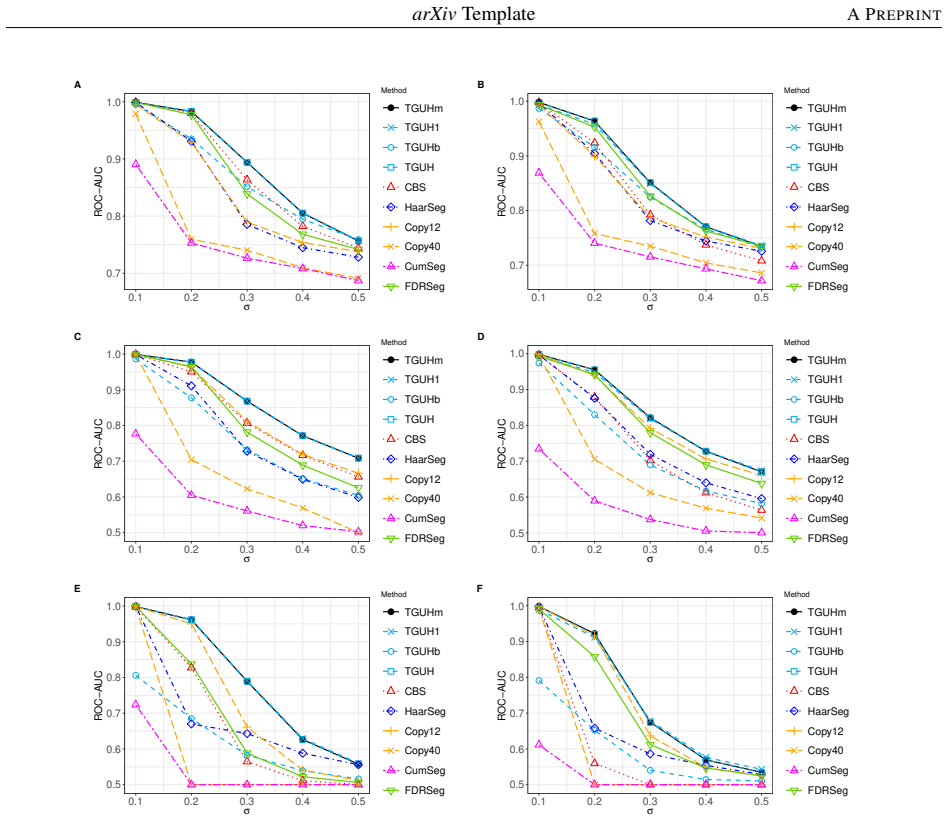

read the original abstract
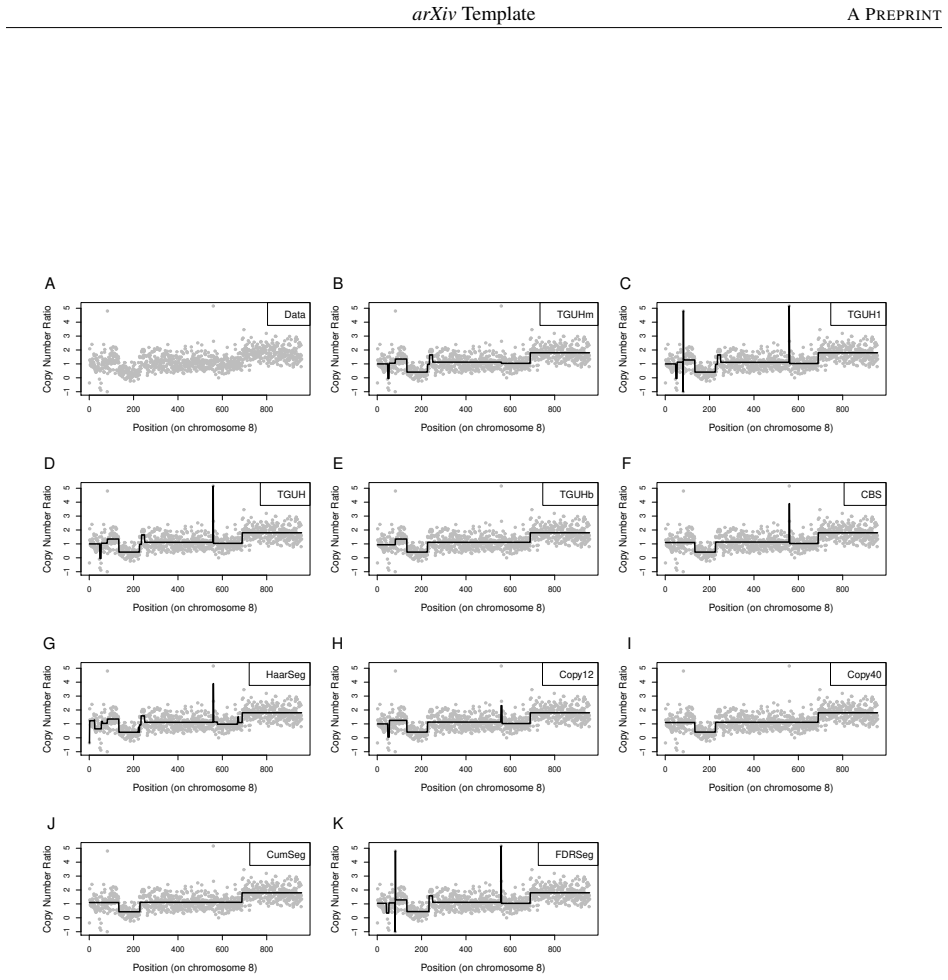
Detecting copy number alterations (CNAs) from next-generation sequencing data remains challenging, particularly for short segments under noisy conditions. Existing segmentation methods often suffer from high false positive rates or fail to reliably detect short aberrations, especially in low-coverage data. In this study, we propose a modified tail-greedy unbalanced Haar (TGUHm) method that introduces a dual-thresholding strategy to improve segmentation accuracy. The proposed approach effectively suppresses spurious spikes while preserving sensitivity to both short and long CNA segments. Extensive simulation studies under Gaussian and heavy-tailed noise demonstrate that TGUHm consistently achieves higher true positive rates and lower false positive rates compared to state-of-the-art methods, including CBS, HaarSeg, and FDRSeg. In particular, the proposed method improves detection accuracy for short segments while maintaining competitive overall performance. Application to real cancer genomic data further confirms the practical utility of the method, revealing biologically meaningful CNAs associated with known cancer-related genes. These results suggest that TGUHm provides a robust and effective framework for CNA detection in challenging sequencing settings.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a modified tail-greedy unbalanced Haar (TGUHm) wavelet segmentation method for detecting copy number alterations (CNAs) in next-generation sequencing data. The key innovation is a dual-thresholding strategy designed to reduce false positives from spurious spikes while maintaining sensitivity to short and long CNA segments. Through simulations under Gaussian and heavy-tailed noise, the authors claim superior true positive rates (TPR) and false positive rates (FPR) compared to CBS, HaarSeg, and FDRSeg, with particular improvements for short segments. The method is also applied to a real cancer dataset to identify biologically relevant CNAs.
Significance. If the claimed performance gains hold under broader conditions, TGUHm could provide a useful refinement for CNA detection in noisy or low-coverage NGS settings, where short segments are particularly difficult to resolve. The adaptation of tail-greedy unbalanced Haar wavelets targets a known limitation of standard wavelet and segmentation approaches. The simulation design covering two noise families and the real-data example are positive elements, but the lack of quantitative performance metrics, error bars, and parameter robustness checks limits the immediate impact.
major comments (3)
- [Abstract] Abstract: the claim that TGUHm 'consistently achieves higher true positive rates and lower false positive rates' is presented without numerical effect sizes, confidence intervals, or any description of how the dual-threshold values were selected or validated, which is load-bearing for the central performance assertion.
- [Simulation studies] Simulation studies: no sensitivity analysis, cross-validation, or justification is given for the specific dual-threshold parameters; this directly affects whether the reported TPR/FPR advantages generalize beyond the exact simulated Gaussian and heavy-tailed models used for evaluation.
- [Real data application] Real-data application: only a single cancer dataset is analyzed, with no reported details on data exclusion rules, preprocessing steps, or multiple-testing correction, which weakens the support for the claim of 'practical utility' and 'biologically meaningful CNAs'.
minor comments (2)
- [Method] The manuscript would benefit from explicit equations defining the dual-thresholding rule and the precise form of the modified tail-greedy step.
- [Figures] Simulation figures should include error bars or variability measures across replicates to allow readers to assess the stability of the reported TPR/FPR differences.
Simulated Author's Rebuttal
We thank the referee for their thoughtful comments, which have helped us identify areas for improvement in the presentation of our results. We address each major comment in turn and outline the revisions we intend to make to the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that TGUHm 'consistently achieves higher true positive rates and lower false positive rates' is presented without numerical effect sizes, confidence intervals, or any description of how the dual-threshold values were selected or validated, which is load-bearing for the central performance assertion.
Authors: We agree that the abstract would benefit from more specific quantitative support for the performance claims. In the revised manuscript, we will update the abstract to include key numerical results from the simulations, such as the TPR and FPR values achieved for short and long segments under both noise models. We will also briefly describe the procedure used to select the dual-threshold values. Full details on these aspects, including any available measures of variability across simulation replicates, will be expanded in the simulation studies section. revision: yes
-
Referee: [Simulation studies] Simulation studies: no sensitivity analysis, cross-validation, or justification is given for the specific dual-threshold parameters; this directly affects whether the reported TPR/FPR advantages generalize beyond the exact simulated Gaussian and heavy-tailed models used for evaluation.
Authors: The specific dual-threshold parameters were determined through preliminary simulation experiments aimed at balancing detection sensitivity and specificity, though this process was not fully detailed in the original submission. We concur that a sensitivity analysis would better demonstrate the robustness of our findings. Accordingly, we will add a sensitivity analysis in the revised version, including plots or tables showing how TPR and FPR vary with different threshold settings under the Gaussian and heavy-tailed noise models. This will help confirm that the advantages over competing methods are not overly sensitive to the exact parameter choices. revision: yes
-
Referee: [Real data application] Real-data application: only a single cancer dataset is analyzed, with no reported details on data exclusion rules, preprocessing steps, or multiple-testing correction, which weakens the support for the claim of 'practical utility' and 'biologically meaningful CNAs'.
Authors: The real-data example is intended as an illustration of the method's application rather than a comprehensive validation study. We will revise this section to include detailed descriptions of the data preprocessing steps, exclusion criteria applied to the cancer dataset, and our approach to multiple-testing (which relies on the method's thresholding mechanism). While expanding to additional datasets would further support generalizability, we believe the current analysis, with these added details, adequately demonstrates the practical utility in identifying biologically relevant CNAs. revision: partial
Circularity Check
No circularity: algorithmic modification evaluated on separate simulations and real data
full rationale
The paper presents TGUHm as an algorithmic extension of the tail-greedy unbalanced Haar wavelet method, introducing a dual-thresholding strategy to suppress spurious spikes while retaining sensitivity to short and long segments. No equations, derivations, or first-principles results are shown that reduce claimed performance metrics to quantities fitted from the same data or defined in terms of the target outputs. Performance claims rest on separate simulation studies (Gaussian and heavy-tailed noise) and application to one real cancer dataset, with comparisons to CBS, HaarSeg, and FDRSeg. Any prior citations to unbalanced Haar work are external to the present modifications and do not form a self-referential chain that forces the reported TPR/FPR improvements. The derivation chain is therefore self-contained and non-circular.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data
Arief Gusnanto, Henry Wood, Yudi Pawitan, Pamela Rabbitts, and Stefano Berri. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics, 28 0 (1): 0 40--47, 2012
2012
-
[2]
Circular binary segmentation for the analysis of array-based dna copy number data
AB Olshen, ES Venkatraman, Robert Lucito, and Michael Wigler. Circular binary segmentation for the analysis of array-based dna copy number data. Biostatistics, 5 0 (4): 0 557--572, 2004
2004
-
[3]
Efficient change point detection for genomic sequences of continuous measurements
Vito Muggeo and Giada Adelfio. Efficient change point detection for genomic sequences of continuous measurements. Bioinformatics, 27 0 (2): 0 161--166, 2010
2010
-
[4]
A fast and flexible method for the segmentation of acgh data
Erez Ben-Yaacov and Yonina C Eldar. A fast and flexible method for the segmentation of acgh data. Bioinformatics, 24 0 (16): 0 i139--i145, 2008
2008
-
[5]
Fdr-control in multiscale change-point segmentation
Housen Li, Axel Munk, and Hannes Sieling. Fdr-control in multiscale change-point segmentation. Electronic Journal of Statistics, 10 0 (1): 0 918--959, 2016
2016
-
[6]
Copynumber: Efficient algorithms for single- and multi-track copy number segmentation
Gro Nilsen, Knut Liestøl, Peter Van Loo, Hans Kristian Moen Vollan, Marianne B Eide, Oscar M Rueda, Suet-Feung Chin, Roslin Russell, Lars O Baumbusch, Carlos Caldas, Anne-Lise Børresen-Dale, and Ole Christian Lingjaerde. Copynumber: Efficient algorithms for single- and multi-track copy number segmentation. BMC genomics, 13 0 (1): 0 591--591, 2012
2012
-
[7]
Comparative analysis of algorithms for identifying amplifications and deletions in array cgh data
Weil R Lai, Mark D Johnson, Raju Kucherlapati, and Peter J Park. Comparative analysis of algorithms for identifying amplifications and deletions in array cgh data. Bioinformatics, 21 0 (19): 0 3763--3770, 2005
2005
-
[8]
A comparison study: Applying segmentation to array CGH data for downstream analyses
Hanni Willenbrock and Jane Fridlyand. A comparison study: Applying segmentation to array CGH data for downstream analyses. Bioinformatics, 21 0 (22): 0 4084--4091, 2005
2005
-
[9]
Using next-generation sequencing for high resolution multiplex analysis of copy number variation from nanogram quantities of DNA from formalin-fixed paraffin-embedded specimens
HM Wood, O Belvedere, C Conway, C Daly, R Chalkley, M Bickerdike, C McKinley, P Egan, L Ross, B Hayward, J Morgan, L Davidson, K MacLennan, TK Ong, K Papagiannopoulos, I Cook, DJ Adams, GR Taylor, and P Rabbitts. Using next-generation sequencing for high resolution multiplex analysis of copy number variation from nanogram quantities of DNA from formalin-f...
2010
-
[10]
Estimating optimal window size for analysis of low-coverage next-generation sequence data
Arief Gusnanto, Charles C Taylor, Ibrahim Nafisah, Henry M Wood, Pamela Rabbitts, and Stefano Berri. Estimating optimal window size for analysis of low-coverage next-generation sequence data. Bioinformatics, 30 0 (13): 0 1823--1829, 2014
2014
-
[11]
Tail-greedy bottom-up data decompositions and fast multiple change-point detection
P Fryzlewicz. Tail-greedy bottom-up data decompositions and fast multiple change-point detection. Annals of Statistics, 46 0 (6B): 0 3390--3421, 2018
2018
-
[12]
A computational index derived from whole-genome copy number analysis is a novel tool for prognosis in early stage lung squamous cell carcinoma
Ornella Belvedere, Stefano Berri, Rebecca Chalkley, Caroline Conway, Fabio Barbone, Federica Pisa, Kenneth MacLennan, Catherine Daly, Melissa Alsop, Joanne Morgan, Jessica Menis, Peter Tcherveniakov, Kostas Papagiannopoulos, Pamela Rabbitts, and Henry M Wood. A computational index derived from whole-genome copy number analysis is a novel tool for prognosi...
2012
-
[13]
Fast and accurate short read alignment with burrows–wheeler transform
Heng Li and Richard Durbin. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics, 25 0 (14): 0 1754--1760, 2009
2009
-
[14]
Assembly of microarrays for genome-wide measurement of dna copy number
Antoine Snijders, Norma Nowak, Richard Segraves, Stephanie Blackwood, Nils Brown, Jeffrey Conroy, Greg Hamilton, Anna Hindle, Bing Huey, Karen Kimura, Sindy Law, Ken Myambo, Joel Palmer, Bauke Ylstra, Jingzhu Yue, Joe Gray, Ajay Jain, Daniel Pinkel, and Donna Albertson. Assembly of microarrays for genome-wide measurement of dna copy number. Nature Genetic...
2001
-
[15]
Gistic2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers
Craig H Mermel, Steven E Schumacher, Barbara Hill, Matthew L Meyerson, Rameen Beroukhim, and Gad Getz. Gistic2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. BioMed Central Ltd, 12 0 (4): 0 R41--R41, 2011
2011
-
[16]
A survey of sampling from contaminated distributions
JW Tukey. A survey of sampling from contaminated distributions. In Contributions to Probability and Statistics: Essays in Honor of Harold Hotelling, pages 448--485, 1960
1960
-
[17]
Performance evaluation of dna copy number segmentation methods
Morgane Pierre-Jean, Guillem Rigaill, and Pierre Neuvial. Performance evaluation of dna copy number segmentation methods. Briefings in Bioinformatics, 16 0 (4): 0 600--615, 2015
2015
-
[18]
Fiona Cunningham, James E Allen, Jamie Allen, Jorge Alvarez-Jarreta, M Ridwan Amode, Irina M Armean, Olanrewaju Austine-Orimoloye, Andrey G Azov, If Barnes, Ruth Bennett, Andrew Berry, Jyothish Bhai, Alexandra Bignell, Konstantinos Billis, Sanjay Boddu, Lucy Brooks, Mehrnaz Charkhchi, Carla Cummins, Luca Da Rin Fioretto, Claire Davidson, Kamalkumar Dodiya...
-
[19]
Hiemstra
P.S. Hiemstra. Defensins. In Geoffrey J. Laurent and Steven D. Shapiro, editors, Encyclopedia of Respiratory Medicine, pages 7--10. Academic Press, Oxford, 2006. ISBN 978-0-12-370879-3
2006
-
[20]
Amplification of chromosome 8 genes in lung cancer
Onur Baykara, Burak Bakir, Nur Buyru, Kamil Kaynak, and Nejat Dalay. Amplification of chromosome 8 genes in lung cancer. Journal of cancer, 6 0 (3): 0 270, 2015
2015
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.