Recognition: unknown
CellxPert: Inference-Time MCMC Steering of a Multi-Omics Single-Cell Foundation Model for In-Silico Perturbation
Pith reviewed 2026-05-09 19:24 UTC · model grok-4.3
The pith
CellxPert steers a multi-omics single-cell model at inference time with Metropolis-Hastings sampling to predict gene perturbation responses more reliably than direct token edits.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
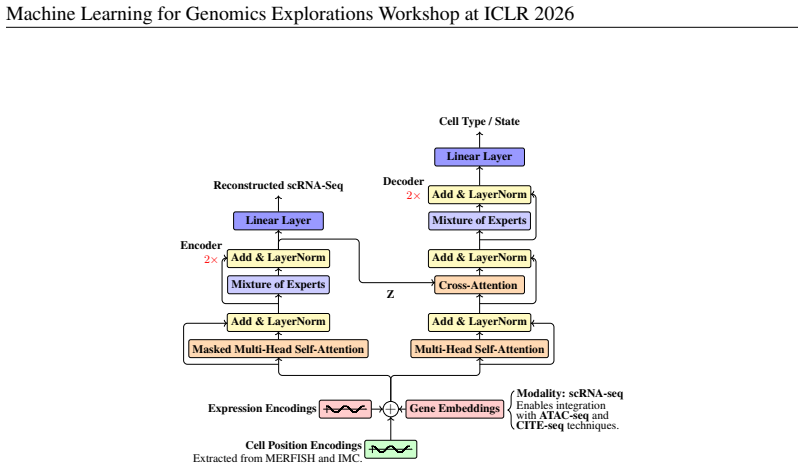
CellxPert is a scalable multimodal foundation model that jointly encodes scRNA-seq, ATAC-seq, CITE-seq and spatial layers from MERFISH or imaging mass-cytometry. For genome-wide transcriptomic response prediction to in-silico perturbations it employs a Metropolis-Hastings sampler whose proposal kernel uses the model's masked conditional distributions to transition to new transcriptomic states conditioned on the perturbed genes. This Markov-chain procedure mitigates out-of-distribution artifacts from abrupt token manipulation and yields biologically interpretable trajectories, leading to better performance than classical and state-of-the-art baselines on PBMC68K, Replogle Perturb-seq, Systema
What carries the argument
Metropolis-Hastings sampler whose proposal kernel uses the model's masked conditional distributions to transition transcriptomic states conditioned on perturbed genes
If this is right
- Surpasses baselines in cell-type annotation across an ontology of 154 largely overlapping identities on PBMC68K and BMMC benchmarks.
- Delivers higher accuracy in genome-wide transcriptomic response prediction to in-silico perturbations on Replogle Perturb-seq and Systema benchmarks.
- Achieves seamless multi-omic integration across transcriptomic, chromatin, proteomic and spatial assays without separate models.
- Supports efficient fine-tuning via Low Rank Adaptation for new downstream tasks while retaining the perturbation steering capability.
- Generates perturbation trajectories that remain within the model's learned distribution rather than jumping to unrealistic states.
Where Pith is reading between the lines
- The same conditional-sampling approach could be applied to other sequence foundation models in biology to replace heuristic editing rules with guided Markov moves.
- Because the method conditions on spatial layers, it may allow perturbation predictions that respect tissue context rather than treating cells in isolation.
- If the trajectories prove interpretable, they could serve as a source of synthetic training data for downstream supervised models of gene regulatory networks.
- Testing on perturbation datasets from different species or disease contexts would reveal whether the MCMC steering generalizes beyond the four evaluated benchmarks.
Load-bearing premise
The Metropolis-Hastings sampler using the model's masked conditional distributions produces biologically interpretable trajectories and mitigates out-of-distribution artifacts introduced by abrupt token manipulation.
What would settle it
A direct comparison on a new held-out Perturb-seq dataset where CellxPert's predicted expression changes after gene knockouts show no closer match to experimental ground truth than simple token deletion or reordering baselines.
Figures







read the original abstract
In this work, we introduce CellxPert, a scalable multimodal foundation model that unifies single-cell and spatial multi-omics within a common representation space. CellxPert jointly encodes transcriptomic (scRNA-seq), chromatin-accessibility (ATAC-seq), and surface-proteomic (CITE-seq) measurements, while directly incorporating MERFISH and imaging mass-cytometry data as 2D or 3D spatial-visual layers. CellxPert facilitates four key downstream tasks out of the box: (i) cell-type annotation across a broad ontology of 154 largely overlapping identities -- the largest label space addressed to date and a stringent test of fine-grained discrimination, (ii) efficient fine-tuning using Low Rank Adaptation (LoRA), (iii) genome-wide transcriptomic response prediction to in-silico perturbations (ISP), and (iv) seamless multi-omic integration across various assays and platforms. Unlike current single-cell foundation models, which approximate gene perturbations by deleting or reordering tokenized gene expression ranks, CellxPert employs a Metropolis-Hastings sampler whose proposal kernel uses the model's masked conditional distributions to transition to new transcriptomic states conditioned on the perturbed genes. This Markov-chain procedure mitigates out-of-distribution artifacts introduced by abrupt token manipulation and produces trajectories that are biologically interpretable. Evaluations on PBMC68K, Replogle Perturb-seq, Systema, and BMMC benchmarks show that CellxPert surpasses classical and state-of-the-art baselines in cell-type annotation, perturbation response prediction, and multi-omic integration.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces CellxPert, a scalable multimodal foundation model unifying single-cell (scRNA-seq, ATAC-seq, CITE-seq) and spatial (MERFISH, imaging mass-cytometry) multi-omics data in a shared representation. It supports four tasks: cell-type annotation over an ontology of 154 largely overlapping identities, LoRA-based fine-tuning, genome-wide transcriptomic response prediction to in-silico perturbations, and multi-omic integration. Unlike prior models that use abrupt gene token deletion or reordering for perturbations, CellxPert employs a Metropolis-Hastings MCMC sampler whose proposals draw from the model's masked conditional distributions to generate new transcriptomic states. This is claimed to reduce out-of-distribution artifacts and yield biologically interpretable trajectories. The model is evaluated on PBMC68K, Replogle Perturb-seq, Systema, and BMMC benchmarks, where it is reported to surpass classical and state-of-the-art baselines in annotation accuracy, perturbation response prediction, and integration performance.
Significance. If the benchmark superiority and MCMC trajectory claims hold with rigorous validation, this represents a meaningful technical step forward for single-cell foundation models. The shift from heuristic token edits to MCMC steering conditioned on the model's own distributions offers a more principled mechanism for in-silico perturbations, potentially improving reliability for downstream applications such as virtual screening. The scale of the label space (154 identities) and the native handling of spatial layers also address practical gaps in current tools, provided the empirical gains are substantiated with quantitative metrics and controls.
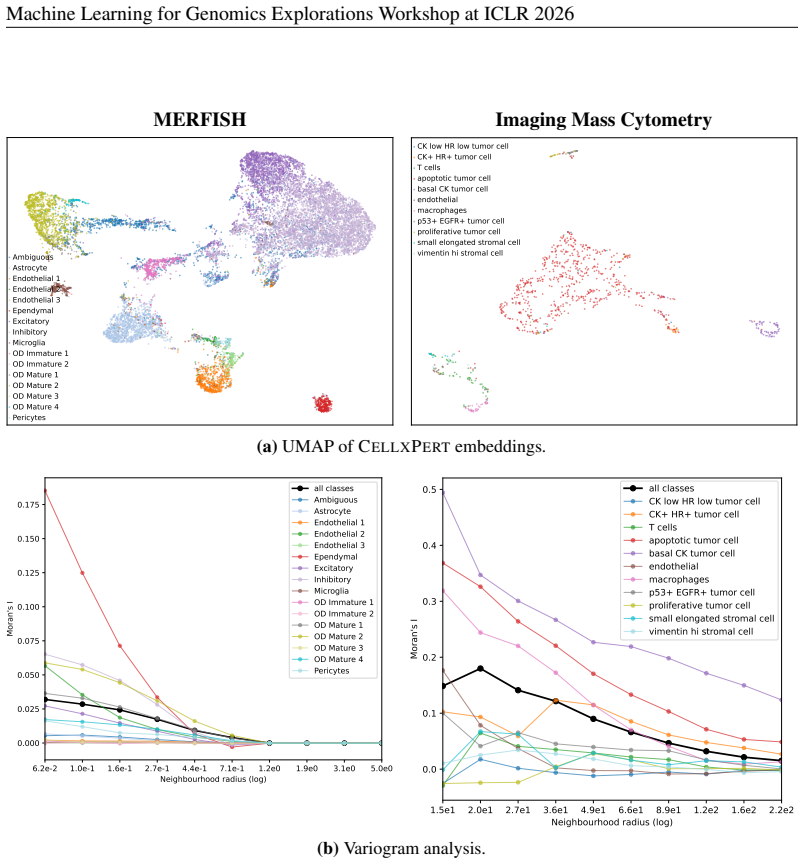
major comments (2)
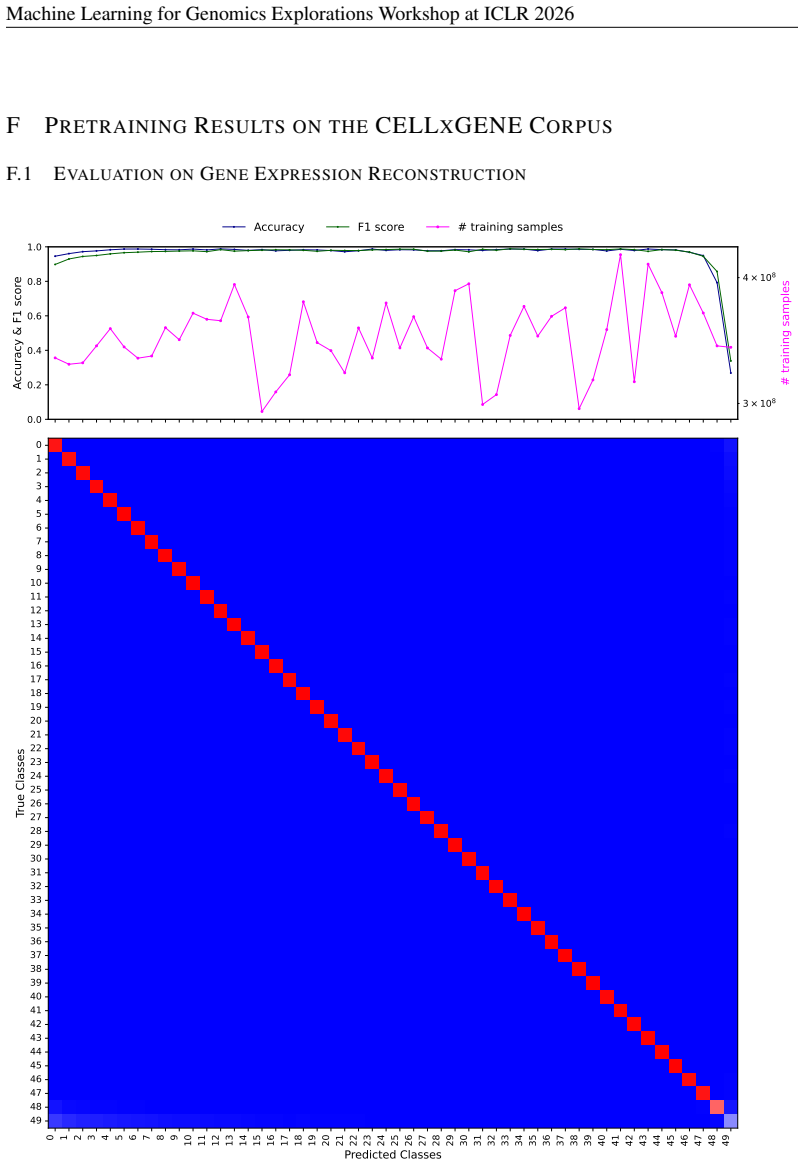
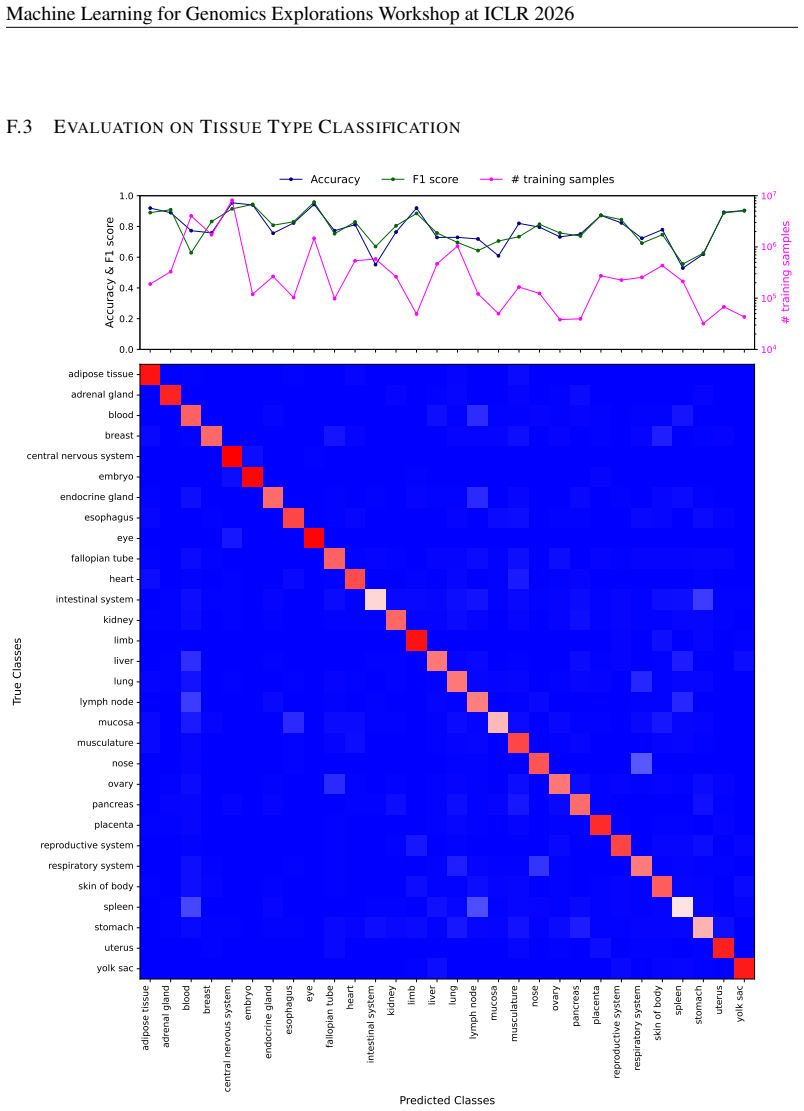
- [Results] Results section (benchmark evaluations): The central claim that CellxPert surpasses baselines on PBMC68K, Replogle Perturb-seq, Systema, and BMMC is load-bearing, yet the manuscript provides no numerical performance values (accuracy, correlation, AUROC, etc.), error bars, or statistical significance tests for any task. Without these, the superiority assertion cannot be assessed and must be supported by explicit tables or figures with all metrics and controls.
- [Methods] Methods (MCMC steering procedure): The Metropolis-Hastings sampler is described as using masked conditional distributions to transition transcriptomic states and mitigate OOD artifacts, but the acceptance probability formula, proposal kernel details, burn-in/convergence criteria, and any diagnostics for trajectory biological plausibility are not specified. This directly affects the validity of the interpretability claim and requires explicit equations or pseudocode.
minor comments (2)
- [Abstract] Abstract: The four downstream tasks are listed but the perturbation task description could explicitly contrast the MCMC approach with the 'deleting or reordering tokenized gene expression ranks' baseline for immediate clarity.
- [Introduction] Notation: The term 'ISP' is introduced without expansion on first use; while inferable from context, an explicit definition would aid readability.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback and for highlighting areas where the manuscript can be strengthened. We address each major comment below and will revise the manuscript to incorporate the requested details, ensuring all claims are rigorously supported.
read point-by-point responses
-
Referee: [Results] Results section (benchmark evaluations): The central claim that CellxPert surpasses baselines on PBMC68K, Replogle Perturb-seq, Systema, and BMMC is load-bearing, yet the manuscript provides no numerical performance values (accuracy, correlation, AUROC, etc.), error bars, or statistical significance tests for any task. Without these, the superiority assertion cannot be assessed and must be supported by explicit tables or figures with all metrics and controls.
Authors: We agree that explicit numerical metrics, error bars, and statistical tests are necessary to substantiate the performance claims. The current version presents results primarily through figures without accompanying tables of exact values or significance testing. In the revised manuscript, we will add a dedicated results table (or expanded supplementary tables) reporting all key metrics—accuracy, Pearson/Spearman correlations, AUROC, etc.—for each benchmark and task, including means and standard deviations across replicates, and p-values from appropriate statistical tests (e.g., paired t-tests or Wilcoxon rank-sum tests) against baselines. Relevant figures will be updated with error bars, and all superiority statements will be qualified by these quantitative results. revision: yes
-
Referee: [Methods] Methods (MCMC steering procedure): The Metropolis-Hastings sampler is described as using masked conditional distributions to transition transcriptomic states and mitigate OOD artifacts, but the acceptance probability formula, proposal kernel details, burn-in/convergence criteria, and any diagnostics for trajectory biological plausibility are not specified. This directly affects the validity of the interpretability claim and requires explicit equations or pseudocode.
Authors: We acknowledge that the MCMC procedure description lacks the necessary technical specificity. In the revised Methods section, we will provide: the full Metropolis-Hastings acceptance probability formula; a precise mathematical definition of the proposal kernel derived from the model's masked conditional distributions; explicit parameters for burn-in, sampling iterations, and convergence diagnostics (e.g., trace plots, effective sample size, or Gelman-Rubin statistic); and additional validation steps for trajectory plausibility, such as gene-set enrichment analysis or alignment with known perturbation responses from the literature. Pseudocode for the full sampler will be included as an algorithm box. revision: yes
Circularity Check
No significant circularity detected
full rationale
The provided abstract and description introduce CellxPert with a Metropolis-Hastings sampler using masked conditional distributions for in-silico perturbations. No equations, fitted parameters renamed as predictions, self-citations as load-bearing premises, or ansatzes smuggled via prior work are present in the given text. The method is described as distinct from abrupt token edits, with performance claims tied to external benchmarks (PBMC68K, Replogle Perturb-seq, etc.) rather than internal redefinitions. The derivation chain is self-contained against the stated inputs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The model's masked conditional distributions accurately capture biological transcriptomic states for use in MCMC proposals
Reference graph
Works this paper leans on
-
[1]
Foundation models meet imbalanced single-cell data when learning cell type annotations.bioRxiv, pp
Abdel Rahman Alsabbagh, Alberto Maillo Ruiz de Infante, David Gomez-Cabrero, Narsis A Kiani, Sumeer Ahmad Khan, and Jesper N Tegnér. Foundation models meet imbalanced single-cell data when learning cell type annotations.bioRxiv, pp. 2023–10,
2023
-
[2]
Data matrix normalization and merging strategies minimize batch-specific systemic variation in scrna-seq data.bioRxiv, pp
Benjamin R Babcock, Astrid Kosters, Junkai Yang, Mackenzie L White, and Eliver EB Ghosn. Data matrix normalization and merging strategies minimize batch-specific systemic variation in scrna-seq data.bioRxiv, pp. 2021–08,
2021
-
[3]
Charlotte Bunne, Yusuf Roohani, Yanay Rosen, Ankit Gupta, Xikun Zhang, Marcel Roed, Theo Alexandrov, Mohammed AlQuraishi, Patricia Brennan, Daniel B Burkhardt, et al. How to build the virtual cell with artificial intelligence: Priorities and opportunities.arXiv preprint arXiv:2409.11654,
-
[4]
Alexis Chevalier, Soumya Ghosh, Urvi Awasthi, James Watkins, Julia Bieniewska, Nichita Mitrea, Olga Kotova, Kirill Shkura, Andrew Noble, Michael Steinbaugh, et al. Teddy: A family of foundation models for understanding single cell biology.arXiv preprint arXiv:2503.03485,
-
[5]
FlashAttention-2: Faster Attention with Better Parallelism and Work Partitioning
11 Machine Learning for Genomics Explorations Workshop at ICLR 2026 Tri Dao. Flashattention-2: Faster attention with better parallelism and work partitioning.arXiv preprint arXiv:2307.08691,
work page internal anchor Pith review arXiv 2026
-
[6]
Bert: Pre-training of deep bidirectional transformers for language understanding
Jacob Devlin, Ming-Wei Chang, Kenton Lee, and Kristina Toutanova. Bert: Pre-training of deep bidirectional transformers for language understanding. InProceedings of the 2019 conference of the North American chapter of the association for computational linguistics: human language technologies, volume 1 (long and short papers), pp. 4171–4186,
2019
-
[7]
Training Compute-Optimal Large Language Models
Jordan Hoffmann, Sebastian Borgeaud, Arthur Mensch, Elena Buchatskaya, Trevor Cai, Eliza Rutherford, Diego de Las Casas, Lisa Anne Hendricks, Johannes Welbl, Aidan Clark, et al. Training compute-optimal large language models.arXiv preprint arXiv:2203.15556,
work page internal anchor Pith review arXiv
-
[8]
Generating novel protein sequences using gibbs sampling of masked language models.bioRxiv, pp
Sean R Johnson, Sarah Monaco, Kenneth Massie, and Zaid Syed. Generating novel protein sequences using gibbs sampling of masked language models.bioRxiv, pp. 2021–01,
2021
-
[9]
Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in parkinson’s disease.Nature neuroscience, 25(5):588–595,
12 Machine Learning for Genomics Explorations Workshop at ICLR 2026 Tushar Kamath, Abdulraouf Abdulraouf, SJ Burris, Jonah Langlieb, Vahid Gazestani, Naeem M Nadaf, Karol Balderrama, Charles Vanderburg, and Evan Z Macosko. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in parkinson’s disease.Na...
2026
-
[10]
Scaling Laws for Neural Language Models
Jared Kaplan, Sam McCandlish, Tom Henighan, Tom B Brown, Benjamin Chess, Rewon Child, Scott Gray, Alec Radford, Jeffrey Wu, and Dario Amodei. Scaling laws for neural language models. arXiv preprint arXiv:2001.08361,
work page internal anchor Pith review Pith/arXiv arXiv 2001
-
[11]
The Power of Scale for Parameter-Efficient Prompt Tuning
Brian Lester, Rami Al-Rfou, and Noah Constant. The power of scale for parameter-efficient prompt tuning.arXiv preprint arXiv:2104.08691,
work page internal anchor Pith review arXiv
-
[12]
B., Hoffer, E., and Reichart, R
Itay Nakash, Nitay Calderon, Eyal Ben David, Elad Hoffer, and Roi Reichart. Adaptivocab: Enhanc- ing llm efficiency in focused domains through lightweight vocabulary adaptation.arXiv preprint arXiv:2503.19693,
-
[13]
A cross-species generative cell atlas across 1.5 billion years of evolution: The transcriptformer single-cell model.bioRxiv, pp
James D Pearce, Sara E Simmonds, Gita Mahmoudabadi, Lakshmi Krishnan, Giovanni Palla, Ana- Maria Istrate, Alexander Tarashansky, Benjamin Nelson, Omar Valenzuela, Donghui Li, et al. A cross-species generative cell atlas across 1.5 billion years of evolution: The transcriptformer single-cell model.bioRxiv, pp. 2025–04,
2025
-
[14]
Vasculo-neuronal coupling and neurovascular coupling at the neurovascular unit: impact of hypertension.Frontiers in physiology, 11:584135,
13 Machine Learning for Genomics Explorations Workshop at ICLR 2026 Jessica L Presa, Flavia Saravia, Zsolt Bagi, and Jessica A Filosa. Vasculo-neuronal coupling and neurovascular coupling at the neurovascular unit: impact of hypertension.Frontiers in physiology, 11:584135,
2026
-
[15]
Comprehensive integration of single-cell data.cell, 177(7):1888–1902,
Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M Mauck, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija. Comprehensive integration of single-cell data.cell, 177(7):1888–1902,
1902
-
[16]
URLhttps://doi.org/10.1038/s41587-025-02777-8
doi: 10.1038/ s41587-025-02777-8. URLhttps://doi.org/10.1038/s41587-025-02777-8. Isaac Virshup, Sergei Rybakov, Fabian J Theis, Philipp Angerer, and F Alexander Wolf. anndata: Annotated data.BioRxiv, pp. 2021–12,
-
[17]
Alex Wang and Kyunghyun Cho. Bert has a mouth, and it must speak: Bert as a markov random field language model.arXiv preprint arXiv:1902.04094,
-
[18]
Cellplm: pre-training of cell language model beyond single cells.BioRxiv, pp
Hongzhi Wen, Wenzhuo Tang, Xinnan Dai, Jiayuan Ding, Wei Jin, Yuying Xie, and Jiliang Tang. Cellplm: pre-training of cell language model beyond single cells.BioRxiv, pp. 2023–10,
2023
-
[19]
Scanpy: large-scale single-cell gene expression data analysis.Genome biology, 19:1–5,
14 Machine Learning for Genomics Explorations Workshop at ICLR 2026 F Alexander Wolf, Philipp Angerer, and Fabian J Theis. Scanpy: large-scale single-cell gene expression data analysis.Genome biology, 19:1–5,
2026
-
[20]
Yan Wu, Esther Wershof, Sebastian M Schmon, Marcel Nassar, Bła˙zej Osi´nski, Ridvan Eksi, Zichao Yan, Rory Stark, Kun Zhang, and Thore Graepel. Perturbench: Benchmarking machine learning models for cellular perturbation analysis.arXiv preprint arXiv:2408.10609,
-
[21]
Tahoe-100m: A giga-scale single-cell perturbation atlas for context-dependent gene function and cellular modeling.bioRxiv, pp
Jesse Zhang, Airol A Ubas, Richard de Borja, Valentine Svensson, Nicole Thomas, Neha Thakar, Ian Lai, Aidan Winters, Umair Khan, Matthew G Jones, et al. Tahoe-100m: A giga-scale single-cell perturbation atlas for context-dependent gene function and cellular modeling.bioRxiv, pp. 2025–02,
2025
-
[22]
15 Machine Learning for Genomics Explorations Workshop at ICLR 2026 A HIERARCHICALABSTRACTIONS INTO ASHAREDLATENTSPACE Model Abstraction Molecular layer.RNA, DNA, and protein sequences are serialized into a flat sequence of tokens and processed with a stack of transformer layers. Although small molecule vocabularies (glycans, lipids, metabolites) are crit...
2026
-
[23]
(with O(n2 d) time complexity, where n is the sequence length and d is the per-head embedding dimension) computationally prohibitive on today’s hardware. Cellular layer.Each omics token, whether it represents a gene, an ATAC -seq k-mer, or a CITE-seq epitope, is mapped twice: first to an identity embedding that captures what the feature is, and second to ...
2017
-
[24]
, K), wheren k is the count of classkin the training set
B.4 FINE-TUNINGOBJECTIVE: CLASS-WEIGHTEDCROSS-ENTROPY Given pooled representationhand logitsW h+bforKclasses, we use inverse-frequency weights αk = 1/nk PK j=1(1/nj) (k= 1, . . . , K), wheren k is the count of classkin the training set. The weighted loss over a batch{(h b, yb)}B b=1 is LwCE =− PB b=1 αyb logp θ(yb |h b) PB b=1 αyb , p θ(· |h) = softmax(W ...
2026
-
[25]
The MoE output is the gated sum of expert outputs (Figure 6): MoE(x) = EX e=1 ge fe(x). B.6 ATTENTIONEFFICIENCY FORLONGCONTEXT We use FlashAttention-v2 to compute exact attention with IO-aware tiling, reducing memory traffic and enabling 4k-token contexts at practical memory/throughput (Dao, 2023). FlashAttention-v2’s customCUDAkernels require GPUs with c...
2023
-
[26]
Thus, for tasks like differential expression analysis, perturbation response prediction and rare cell type detection, binning offers an edge
(rank-based) on imbalanced data with rare cell subpopulations (Alsabbagh et al., 2023). Thus, for tasks like differential expression analysis, perturbation response prediction and rare cell type detection, binning offers an edge. D.2 TOKENIZATION After discretizing expression into B=50 bins, each cell c is represented by twoaligned sequences of length L: ...
2023
-
[27]
If [CLS] is enabled, we set gc,1=V , bc,1=0 (neutral bin), shift the original pairs by one, and take L=G+1 (otherwise L=G)
on training genes to map each symbol to [0, V−1] and reserve the unused index V for [CLS]. If [CLS] is enabled, we set gc,1=V , bc,1=0 (neutral bin), shift the original pairs by one, and take L=G+1 (otherwise L=G). We denote the token sequence byT c = (gc,1, bc,1), . . . ,(gc,L, bc,L) and identifyN:=L≤4096. D.3 EMBEDDINGS ANDINPUTCOMPOSITION Letdbe the em...
2025
-
[28]
Homo sapiens
or prompt tuning (Liu et al., 2023). D.6 MAKEMORE WITHLESSDATA During pretraining we augment dataset size via a novelbootstrap -styleaugmentation technique that replicates each cell with independently permuted gene indices. Bootstrapped Permutation Every cell is cloned four times. Each clone receives an independent random permutation of its 4096 highly va...
2023
-
[29]
writes a checkpoint containing the full model state dict, optimizer and scheduler states, and AMP scaler state to disk. If training is interrupted or relaunched with the use_latest_checkpoint flag, all processes synchronize at startup, load this latest checkpoint, restore epoch/partition counters and hyperparameter schedules exactly as they were, and cont...
2026
-
[30]
The Kamath et al
dataset. The Kamath et al. dataset comprises gene expression profiling data obtained through high-throughput sequencing. This study focused on midbrain dopamine (DA) neurons in the substantia nigra pars compacta (SNpc), which are critical for voluntary movements, reward processing, and working memory, and are highly susceptible to neurodegeneration in Par...
2024
-
[31]
Unlike the centroid, which can be skewed by outliers, the medoid is an actual observed cell that retains realistic co-expression structure and is empirically more robust (Park & Jun, 2009; Bulté & Sørensen, 2024). H.3.2LOGUNNORMALIZEDTARGETDISTRIBUTION Let Mperturbed ={m k}K k=1 be the set of perturbed-class anchors, specifically the top-K medoids from a ...
2009
-
[32]
Its primary limitation is that only a predefined gene set is measured, rather than the full transcriptome
and subcellular spatial resolution (Moffitt et al., 2018), allowing reconstruction of a high-resolution 3D point cloud of cells and their microenvironments. Its primary limitation is that only a predefined gene set is measured, rather than the full transcriptome. For our analysis, we use a subset of the MERFISH dataset comprising 73655 cells from (Moffitt...
2018
-
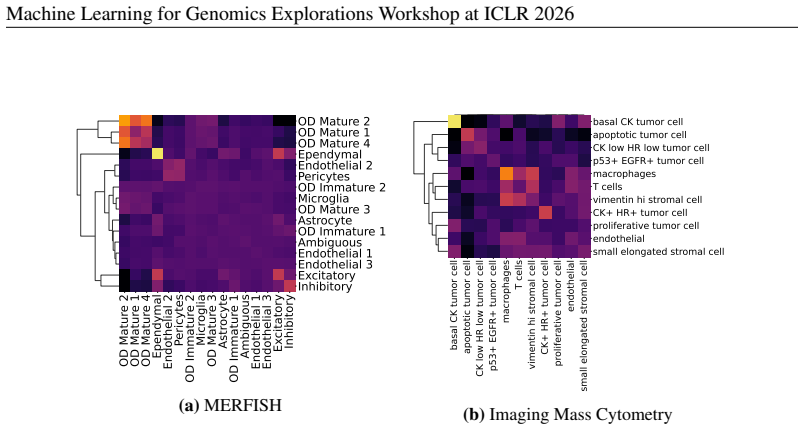
[33]
CK low HR low tumor cell CK+ HR+ tumor cell T cells apoptotic tumor cell basal CK tumor cell endothelial macrophages p53+ EGFR+ tumor cell proliferative tumor cell small elongated stromal cell vimentin hi stromal cell (a)UMAP of CELLXPERTembeddings. 6.2e-2 1.0e-1 1.6e-1 2.7e-1 4.4e-1 7.1e-1 1.2e0 1.9e0 3.1e0 5.0e0 Neighbourhood radius (log) 0.000 0.025 0....
1950
-
[34]
In contrast, Macrophageis depleted near the apoptotic tumor state and apoptotic tumor is depleted relative to major stromal classes
Epithelial tumor classesBasal CKandCK+ HR+show strong self enrichment and only moderate mutual adjacency, which is consistent with contiguous tumor patches rather than one fused epithelial block.Macrophageco-enriches withT Celland withVimentin High Stromal Cell, which marks an immune stromal interface common in solid tumors (Wu et al., 2021; Jackson et al...
2021
-
[35]
a collection of 93 million cells (63% of them are from human), remain constrained by these confounders. Moreover, scaling laws for Large Language Model (LLM) pretraining predict diminishing returns from enlarging model size alone (Hoffmann et al., 2022; Kaplan et al., 2020); hence, synthetic data generation via generative models, in-silico perturbations, ...
2022
-
[36]
proposes complementing the Human Cell Atlas with a Perturbation Cell Atlas to enabletruly causal foundation models. In this landscape, CELLXPERT, despite its modest 26.3 M parameters, achieves a very competitive accuracy by integrating multimodal single-cell signals in an efficient sparse Mixture-of-Experts Transformer, demonstrating that data diversity a...
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.