Recognition: unknown
A-CODE: Fully Atomic Protein Co-Design with Unified Multimodal Diffusion
Pith reviewed 2026-05-09 16:20 UTC · model grok-4.3
The pith
A single unified diffusion process on atoms co-designs protein sequences and structures in one stage.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
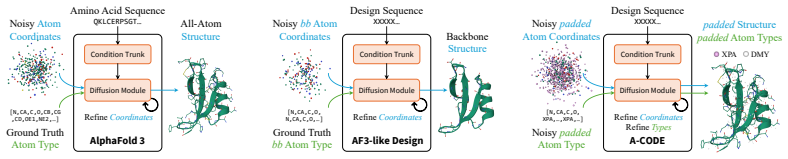
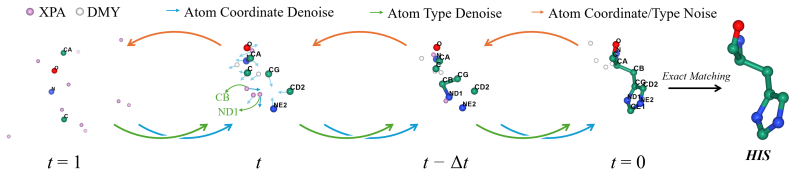
A-CODE is a fully atomic unified one-stage protein co-design model that simultaneously refines discrete atom types and continuous atom coordinates within a multimodal diffusion framework in which residue identities are inferred solely from atom-level predictions. Built on an all-atom architecture, it achieves superior designability for unconditional protein generation over existing one-stage and two-stage models, rivals or outperforms state-of-the-art two-stage models on binder design, and delivers a tenfold improvement in success rate versus the prior one-stage co-design model on hard tasks while enabling seamless non-canonical amino acid modeling.
What carries the argument
The unified multimodal diffusion framework that operates directly on all-atom coordinates and types, with amino acid identities inferred from the atomic predictions.
If this is right
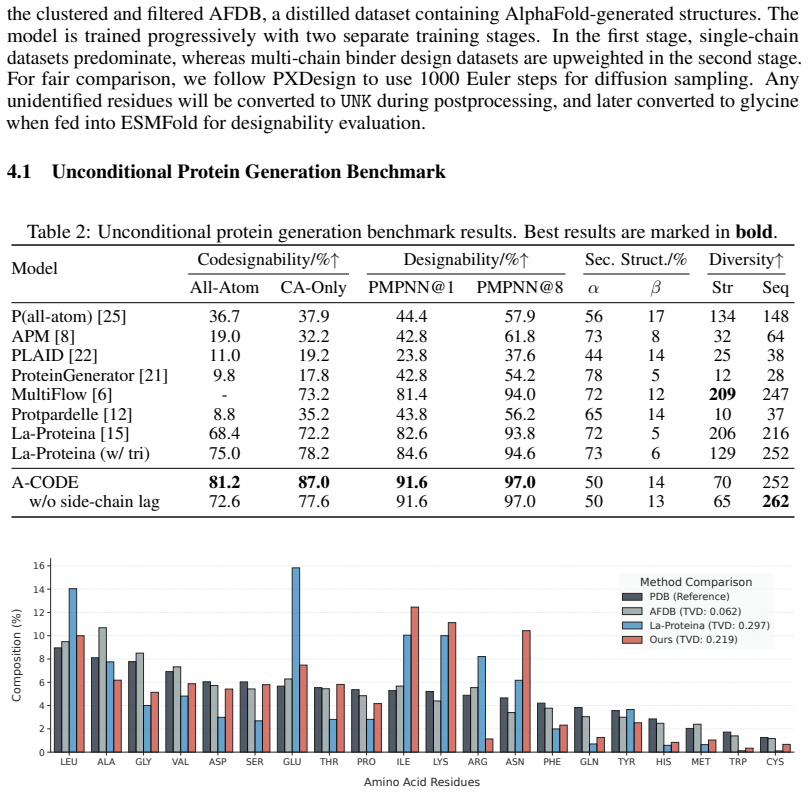
- Unconditional protein generation produces higher designability scores than prior one-stage or two-stage models.
- Binder design achieves success rates up to ten times higher than existing one-stage co-design methods on difficult targets.
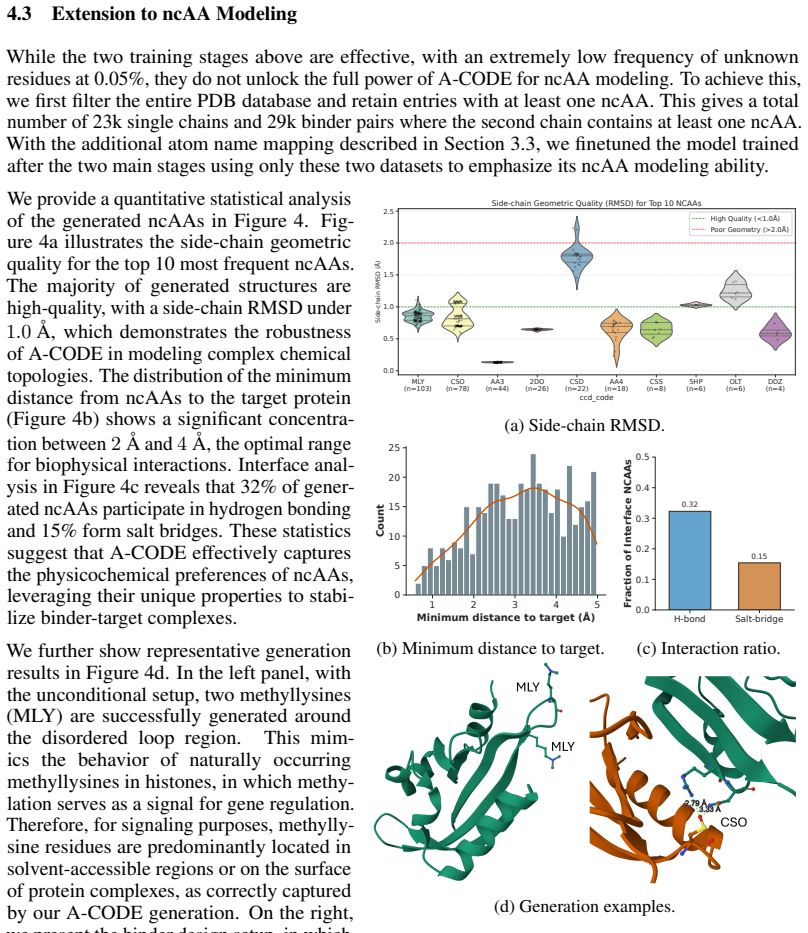
- Non-canonical amino acids can be generated directly inside the same atomic diffusion process.
- The framework provides a single-stage foundation that can extend to more complex biomolecular systems.
Where Pith is reading between the lines
- Removing the separate sequence optimization stage may reduce accumulation of sequence-structure mismatches that occur in cascaded pipelines.
- The same atomic diffusion backbone could be tested on larger multi-domain proteins or on tasks that include explicit ligand atoms.
- Direct atomic modeling might simplify incorporation of post-translational modifications or metal-binding sites without new model components.
Load-bearing premise
Residue identities can be reliably inferred solely from atom-level predictions within the unified diffusion process and the all-atom architecture delivers the reported gains without post-hoc adjustments.
What would settle it
An independent replication on standard protein design benchmarks that measures designability and binder success rates and finds no statistically significant improvement over the strongest two-stage baselines would falsify the central performance claims.
Figures
read the original abstract
We present A-CODE, a fully atomic unified one-stage protein co-design model that simultaneously refines discrete atom types and continuous atom coordinates. Unlike predominant two-stage methods that cascade structure design with amino acid-level sequence design, our approach is fully atomic within a unified multimodal diffusion framework, in which residue identities are inferred solely from atom-level predictions. Built upon the powerful all-atom architecture, A-CODE achieves superior designability for unconditional protein generation, outperforming all existing one-stage and two-stage design models. For binder design, A-CODE rivals and even outperforms existing state-of-the-art two-stage design models and, compared with the existing one-stage co-design model, achieves a drastic tenfold improvement in success rate on hard tasks. The inherent flexibility of our atomic formulation enables, for the first time, seamless adaptation to non-canonical amino acid (ncAA) modeling. Our fully atomic framework establishes a new, versatile foundation for all-atom generative modeling that can be naturally extended to complex biomolecular systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces A-CODE, a unified multimodal diffusion framework for fully atomic one-stage protein co-design. It simultaneously refines discrete atom types and continuous atom coordinates within a single model, with residue identities inferred solely from the atom-level predictions. The work claims superior designability for unconditional protein generation over all prior one-stage and two-stage models, a tenfold improvement in binder-design success rate on hard tasks relative to existing one-stage co-design baselines, and native support for non-canonical amino acid modeling.
Significance. If the performance claims and the strictly one-stage atomic inference procedure are substantiated, the work would offer a meaningful simplification of the protein design pipeline by eliminating cascaded sequence-design stages. The extension to ncAA modeling is a concrete strength that could enable new applications. The all-atom formulation also provides a natural route toward modeling larger biomolecular complexes.
major comments (2)
- [§3.2] §3.2 (Residue identity inference): The procedure for obtaining discrete residue types from predicted atom types and coordinates must be stated explicitly (e.g., direct argmax on atom-type logits, a learned mapping, or any auxiliary classifier). Any unstated post-processing step would undermine the central 'solely from atom-level predictions' and 'one-stage' claims that are used to differentiate A-CODE from two-stage and prior one-stage baselines in §5.
- [§5.2] §5.2 (Binder design results): The reported tenfold success-rate improvement on hard tasks is presented without error bars, trial counts, exact success-rate definition, or ablation of the inference step. These omissions make it impossible to assess whether the gain is statistically robust or driven by the diffusion architecture itself versus hidden sequence-level components.
minor comments (2)
- [Abstract] Abstract: Quantitative metrics, dataset sizes, and pointers to specific result tables are absent, reducing the abstract's utility as a standalone summary.
- [§3.1] Notation: The distinction between atom-type diffusion and coordinate diffusion in the multimodal framework should be clarified with explicit equations or a dedicated diagram panel.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback and the opportunity to clarify key aspects of our work. We address each major comment point-by-point below, with revisions made to enhance clarity and rigor where appropriate.
read point-by-point responses
-
Referee: [§3.2] §3.2 (Residue identity inference): The procedure for obtaining discrete residue types from predicted atom types and coordinates must be stated explicitly (e.g., direct argmax on atom-type logits, a learned mapping, or any auxiliary classifier). Any unstated post-processing step would undermine the central 'solely from atom-level predictions' and 'one-stage' claims that are used to differentiate A-CODE from two-stage and prior one-stage baselines in §5.
Authors: We agree that explicit specification of the inference procedure is essential to substantiate the one-stage atomic claims. In A-CODE, residue identities are obtained directly from the model's atom-level outputs via argmax over the predicted atom-type logits for the atoms belonging to each residue, followed by a deterministic lookup table that maps the resulting atomic composition to the corresponding amino acid identity. No auxiliary classifier, learned mapping, or additional post-processing is applied. We have revised §3.2 to include this description together with pseudocode, ensuring the procedure is fully transparent and reinforcing the distinction from cascaded two-stage approaches. revision: yes
-
Referee: [§5.2] §5.2 (Binder design results): The reported tenfold success-rate improvement on hard tasks is presented without error bars, trial counts, exact success-rate definition, or ablation of the inference step. These omissions make it impossible to assess whether the gain is statistically robust or driven by the diffusion architecture itself versus hidden sequence-level components.
Authors: We acknowledge these omissions limit interpretability. The success rate is defined as the fraction of designs satisfying both structural fidelity (backbone RMSD < 2 Å to the target complex) and functional compatibility (Rosetta binding energy below a task-specific threshold). We have added error bars (standard deviation across 10 independent sampling runs with distinct seeds) and explicit trial counts (100 designs per method per hard task) to the revised §5.2 and supplementary tables. An ablation isolating the atomic diffusion steps (with no sequence-level components present at inference) confirms the performance gain originates from the unified multimodal model rather than hidden post-processing. revision: yes
Circularity Check
No circularity: architecture and performance claims are independent of self-referential inputs.
full rationale
The paper presents A-CODE as a new all-atom diffusion architecture for one-stage protein co-design, with residue identities stated to be inferred from atom-level predictions within the unified framework. No equations, fitted parameters, or derivation steps are exhibited that reduce any claimed result (e.g., designability or binder success rates) to a self-definition, a renamed fit, or a load-bearing self-citation chain. Performance is benchmarked against external prior models rather than internal constructs, and the abstract's contrasts with two-stage and one-stage baselines rely on those external comparisons. The derivation is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Accurate structure prediction of biomolecular interactions with alphafold 3.Nature, 630(8016):493–500, 2024
Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J Ballard, Joshua Bambrick, et al. Accurate structure prediction of biomolecular interactions with alphafold 3.Nature, 630(8016):493–500, 2024
2024
-
[2]
Structured denoising diffusion models in discrete state-spaces.Advances in neural information processing systems, 34:17981–17993, 2021
Jacob Austin, Daniel D Johnson, Jonathan Ho, Daniel Tarlow, and Rianne Van Den Berg. Structured denoising diffusion models in discrete state-spaces.Advances in neural information processing systems, 34:17981–17993, 2021
2021
-
[3]
Beals, L
M. Beals, L. Gross, and S. Harrell. Amino Acid Frequency.https://legacy.nimbios.org/ ~gross/bioed/webmodules/aminoacid.htm, 1999
1999
-
[4]
Avishek Joey Bose, Tara Akhound-Sadegh, Guillaume Huguet, Kilian Fatras, Jarrid Rector- Brooks, Cheng-Hao Liu, Andrei Cristian Nica, Maksym Korablyov, Michael Bronstein, and Alexander Tong. Se (3)-stochastic flow matching for protein backbone generation.arXiv preprint arXiv:2310.02391, 2023
-
[5]
De novo design of all-atom biomolecular interactions with rfdiffusion3.bioRxiv, 2025
Jasper Butcher, Rohith Krishna, Raktim Mitra, Rafael I Brent, Yanjing Li, Nathaniel Corley, Paul T Kim, Jonathan Funk, Simon Mathis, Saman Salike, et al. De novo design of all-atom biomolecular interactions with rfdiffusion3.bioRxiv, 2025
2025
-
[6]
Andrew Campbell, Jason Yim, Regina Barzilay, Tom Rainforth, and Tommi Jaakkola. Gener- ative flows on discrete state-spaces: Enabling multimodal flows with applications to protein co-design.arXiv preprint arXiv:2402.04997, 2024
-
[7]
Flow matching on general geometries.arXiv preprint arXiv:2302.03660, 2023
Ricky TQ Chen and Yaron Lipman. Flow matching on general geometries.arXiv preprint arXiv:2302.03660, 2023
-
[8]
An all-atom generative model for designing protein complexes.arXiv preprint arXiv:2504.13075, 2025
Ruizhe Chen, Dongyu Xue, Xiangxin Zhou, Zaixiang Zheng, Xiangxiang Zeng, and Quan- quan Gu. An all-atom generative model for designing protein complexes.arXiv preprint arXiv:2504.13075, 2025
-
[9]
Molprobity: all-atom structure validation for macromolecular crystallography.Biological crystallography, 66(1):12–21, 2010
Vincent B Chen, W Bryan Arendall, Jeffrey J Headd, Daniel A Keedy, Robert M Immormino, Gary J Kapral, Laura W Murray, Jane S Richardson, and David C Richardson. Molprobity: all-atom structure validation for macromolecular crystallography.Biological crystallography, 66(1):12–21, 2010. 10
2010
-
[10]
Protenix-advancing structure prediction through a comprehensive alphafold3 reproduction.BioRxiv, pages 2025–01, 2025
Xinshi Chen, Yuxuan Zhang, Chan Lu, Wenzhi Ma, Jiaqi Guan, Chengyue Gong, Jincai Yang, Hanyu Zhang, Ke Zhang, et al. Protenix-advancing structure prediction through a comprehensive alphafold3 reproduction.BioRxiv, pages 2025–01, 2025
2025
-
[11]
Categorical flow matching on statistical manifolds.Advances in Neural Information Processing Systems, 37:54787–54819, 2024
Chaoran Cheng, Jiahan Li, Jian Peng, and Ge Liu. Categorical flow matching on statistical manifolds.Advances in Neural Information Processing Systems, 37:54787–54819, 2024
2024
-
[12]
An all-atom protein generative model.Proceedings of the National Academy of Sciences, 121(27):e2311500121, 2024
Alexander E Chu, Jinho Kim, Lucy Cheng, Gina El Nesr, Minkai Xu, Richard W Shuai, and Po-Ssu Huang. An all-atom protein generative model.Proceedings of the National Academy of Sciences, 121(27):e2311500121, 2024
2024
-
[13]
Robust deep learning–based protein sequence design using proteinmpnn.Science, 378(6615):49–56, 2022
Justas Dauparas, Ivan Anishchenko, Nathaniel Bennett, Hua Bai, Robert J Ragotte, Lukas F Milles, Basile IM Wicky, Alexis Courbet, Rob J de Haas, Neville Bethel, et al. Robust deep learning–based protein sequence design using proteinmpnn.Science, 378(6615):49–56, 2022
2022
-
[14]
Discrete flow matching.Advances in Neural Information Processing Systems, 37:133345–133385, 2024
Itai Gat, Tal Remez, Neta Shaul, Felix Kreuk, Ricky TQ Chen, Gabriel Synnaeve, Yossi Adi, and Yaron Lipman. Discrete flow matching.Advances in Neural Information Processing Systems, 37:133345–133385, 2024
2024
-
[15]
Tomas Geffner, Kieran Didi, Zhonglin Cao, Danny Reidenbach, Zuobai Zhang, Christian Dallago, Emine Kucukbenli, Karsten Kreis, and Arash Vahdat. La-proteina: Atomistic protein generation via partially latent flow matching.arXiv preprint arXiv:2507.09466, 2025
-
[16]
Denoising diffusion probabilistic models.Advances in neural information processing systems, 33:6840–6851, 2020
Jonathan Ho, Ajay Jain, and Pieter Abbeel. Denoising diffusion probabilistic models.Advances in neural information processing systems, 33:6840–6851, 2020
2020
-
[17]
Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ron- neberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
2021
-
[18]
Elucidating the design space of diffusion-based generative models.Advances in neural information processing systems, 35: 26565–26577, 2022
Tero Karras, Miika Aittala, Timo Aila, and Samuli Laine. Elucidating the design space of diffusion-based generative models.Advances in neural information processing systems, 35: 26565–26577, 2022
2022
-
[19]
Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
2023
-
[20]
Flow Matching for Generative Modeling
Yaron Lipman, Ricky TQ Chen, Heli Ben-Hamu, Maximilian Nickel, and Matt Le. Flow matching for generative modeling.arXiv preprint arXiv:2210.02747, 2022
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[21]
Multistate and functional protein design using rosettafold sequence space diffusion.Nature biotechnology, 43(8):1288–1298, 2025
Sidney Lyayuga Lisanza, Jacob Merle Gershon, Samuel WK Tipps, Jeremiah Nelson Sims, Lucas Arnoldt, Samuel J Hendel, Miriam K Simma, Ge Liu, Muna Yase, Hongwei Wu, et al. Multistate and functional protein design using rosettafold sequence space diffusion.Nature biotechnology, 43(8):1288–1298, 2025
2025
-
[22]
Generating all-atom protein structure from sequence-only training data.bioRxiv, pages 2024–12, 2024
Amy X Lu, Wilson Yan, Sarah A Robinson, Kevin K Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter Abbeel, and Nathan Frey. Generating all-atom protein structure from sequence-only training data.bioRxiv, pages 2024–12, 2024
2024
-
[23]
Conditional protein structure generation with protpardelle-1c.bioRxiv, 2025
Tianyu Lu, Richard Shuai, Petr Kouba, Zhaoyang Li, Yilin Chen, Akio Shirali, Jinho Kim, and Po-Ssu Huang. Conditional protein structure generation with protpardelle-1c.bioRxiv, 2025
2025
-
[24]
Instructplm: Aligning protein language models to follow protein structure instructions.bioRxiv, pages 2024–04, 2024
Jiezhong Qiu, Junde Xu, Jie Hu, Hanqun Cao, Liya Hou, Zijun Gao, Xinyi Zhou, Anni Li, Xiujuan Li, Bin Cui, et al. Instructplm: Aligning protein language models to follow protein structure instructions.bioRxiv, pages 2024–04, 2024
2024
-
[25]
P (all-atom) is unlocking new path for protein design.bioRxiv, pages 2024–08, 2024
Wei Qu, Jiawei Guan, Rui Ma, Ke Zhai, Weikun Wu, and Haobo Wang. P (all-atom) is unlocking new path for protein design.bioRxiv, pages 2024–08, 2024
2024
-
[26]
Pxdesign: Fast, modular, and accurate de novo design of protein binders.bioRxiv, pages 2025–08, 2025
Milong Ren, Jinyuan Sun, Jiaqi Guan, Cong Liu, Chengyue Gong, Yuzhe Wang, Lan Wang, Qixu Cai, Wenzhi Ma, et al. Pxdesign: Fast, modular, and accurate de novo design of protein binders.bioRxiv, pages 2025–08, 2025. 11
2025
-
[27]
Wiley Online Library, 2001
Michael G Rossmann and Eddy Arnold.International tables for crystallography volume F: crystallography of biological macromolecules. Wiley Online Library, 2001
2001
-
[28]
Simple and effective masked diffusion language models.Advances in Neural Information Processing Systems, 37:130136–130184, 2024
Subham Sahoo, Marianne Arriola, Yair Schiff, Aaron Gokaslan, Edgar Marroquin, Justin Chiu, Alexander Rush, and V olodymyr Kuleshov. Simple and effective masked diffusion language models.Advances in Neural Information Processing Systems, 37:130136–130184, 2024
2024
-
[29]
Score-Based Generative Modeling through Stochastic Differential Equations
Yang Song, Jascha Sohl-Dickstein, Diederik P Kingma, Abhishek Kumar, Stefano Ermon, and Ben Poole. Score-based generative modeling through stochastic differential equations.arXiv preprint arXiv:2011.13456, 2020
work page internal anchor Pith review Pith/arXiv arXiv 2011
-
[30]
Boltzgen: Toward universal binder design.bioRxiv, pages 2025–11, 2025
Hannes Stark, Felix Faltings, MinGyu Choi, Yuxin Xie, Eunsu Hur, Timothy John O’Donnell, Anton Bushuiev, Talip Uçar, Saro Passaro, Weian Mao, et al. Boltzgen: Toward universal binder design.bioRxiv, pages 2025–11, 2025
2025
-
[31]
Fast and accurate protein structure search with foldseek.Nature biotechnology, 42(2):243–246, 2024
Michel Van Kempen, Stephanie S Kim, Charlotte Tumescheit, Milot Mirdita, Jeongjae Lee, Cameron LM Gilchrist, Johannes Söding, and Martin Steinegger. Fast and accurate protein structure search with foldseek.Nature biotechnology, 42(2):243–246, 2024
2024
-
[32]
Diffusion language models are versatile protein learners.arXiv preprint arXiv:2402.18567, 2024
Xinyou Wang, Zaixiang Zheng, Fei Ye, Dongyu Xue, Shujian Huang, and Quanquan Gu. Diffusion language models are versatile protein learners.arXiv preprint arXiv:2402.18567, 2024
-
[33]
Dplm-2: A multimodal diffusion protein language model.arXiv preprint arXiv:2410.13782, 2024
Xinyou Wang, Zaixiang Zheng, Fei Ye, Dongyu Xue, Shujian Huang, and Quanquan Gu. Dplm-2: A multimodal diffusion protein language model.arXiv preprint arXiv:2410.13782, 2024
-
[34]
De novo design of protein structure and function with rfdiffusion.Nature, 620(7976):1089–1100, 2023
Joseph L Watson, David Juergens, Nathaniel R Bennett, Brian L Trippe, Jason Yim, Helen E Eisenach, Woody Ahern, Andrew J Borst, Robert J Ragotte, Lukas F Milles, et al. De novo design of protein structure and function with rfdiffusion.Nature, 620(7976):1089–1100, 2023
2023
-
[35]
Fast protein backbone generation with se (3) flow matching,
Jason Yim, Andrew Campbell, Andrew YK Foong, Michael Gastegger, José Jiménez-Luna, Sarah Lewis, Victor Garcia Satorras, Bastiaan S Veeling, Regina Barzilay, Tommi Jaakkola, et al. Fast protein backbone generation with se (3) flow matching.arXiv preprint arXiv:2310.05297, 2023
-
[36]
Vinicius Zambaldi, David La, Alexander E Chu, Harshnira Patani, Amy E Danson, Tristan OC Kwan, Thomas Frerix, Rosalia G Schneider, David Saxton, Ashok Thillaisundaram, et al. De novo design of high-affinity protein binders with alphaproteo.arXiv preprint arXiv:2409.08022, 2024
-
[37]
Odesign: A world model for biomolecular interaction design.arXiv preprint arXiv:2510.22304, 2025
Odin Zhang, Xujun Zhang, Haitao Lin, Cheng Tan, Qinghan Wang, Yuanle Mo, Qiantai Feng, Gang Du, Yuntao Yu, Zichang Jin, et al. Odesign: A world model for biomolecular interaction design.arXiv preprint arXiv:2510.22304, 2025
-
[38]
A reparameterized discrete diffusion model for text generation.arXiv preprint arXiv:2302.05737,
Lin Zheng, Jianbo Yuan, Lei Yu, and Lingpeng Kong. A reparameterized discrete diffusion model for text generation.arXiv preprint arXiv:2302.05737, 2023. 12 Supplementary Material A Dataset Pipeline In this section, we describe the details of our data pipeline for adapting PXDesign to an all-atom co-design model. Specifically, we discuss our procedure for ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.