Recognition: 2 theorem links
· Lean TheoremBenchmarking Compositional Generalisation for Machine Learning Interatomic Potentials
Pith reviewed 2026-05-12 02:12 UTC · model grok-4.3
The pith
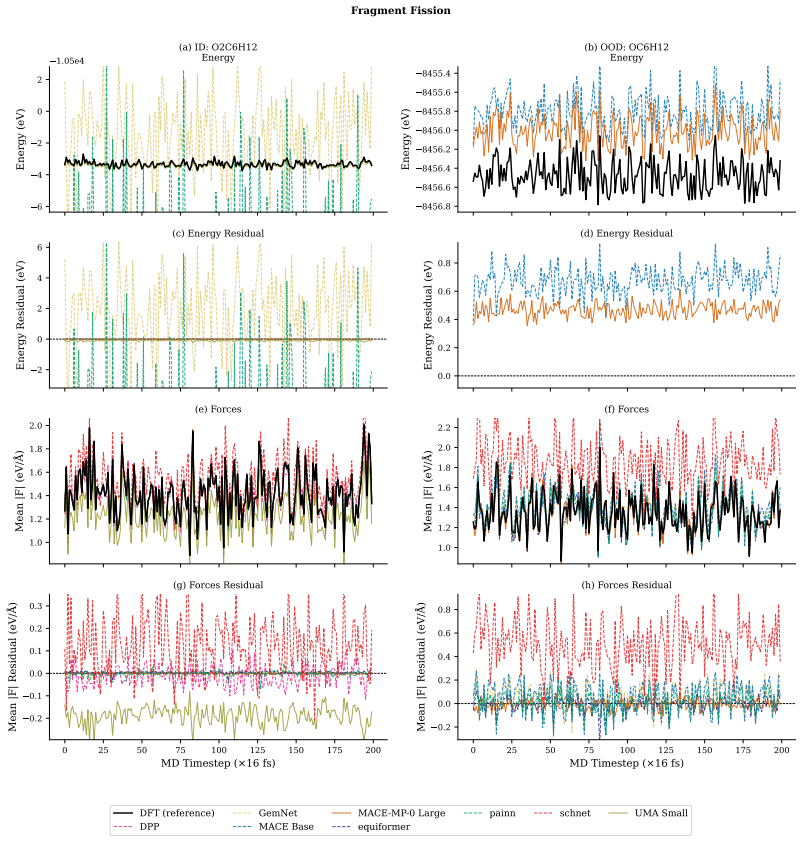
Machine learning interatomic potentials show errors an order of magnitude higher on out-of-distribution molecules than on training examples.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
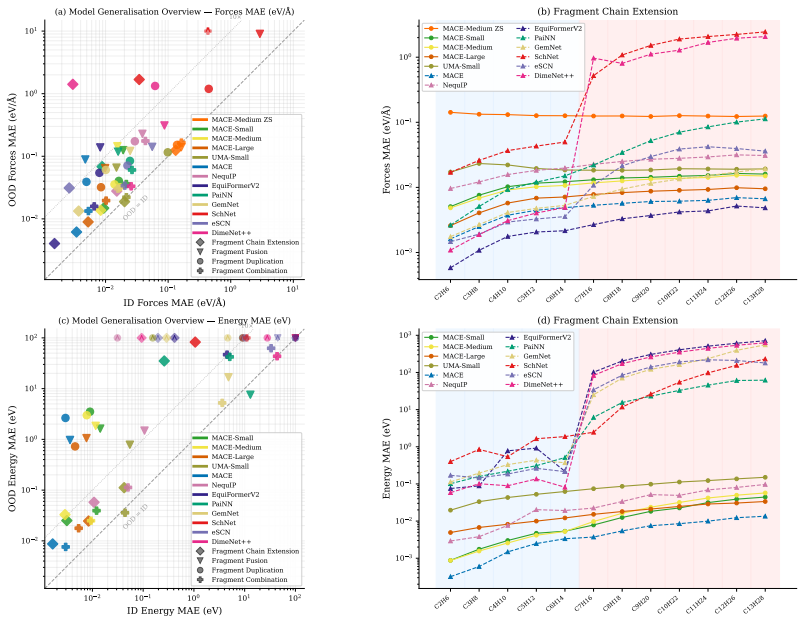
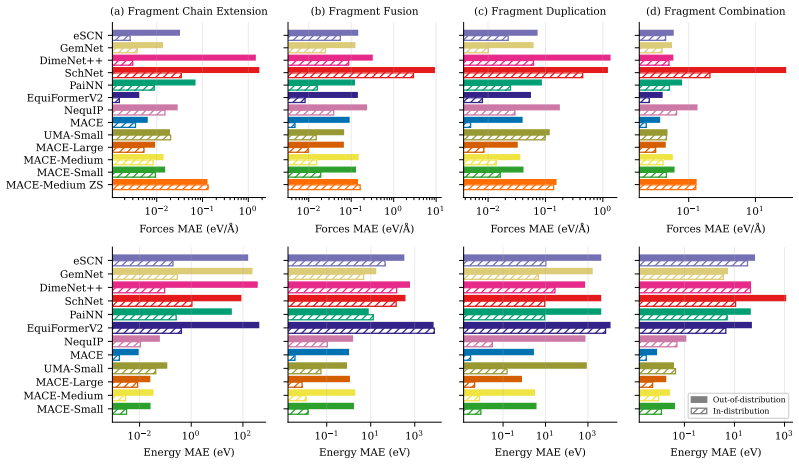
The authors construct a benchmark consisting of four tasks that require compositional generalisation. Models are trained on molecules whose fragments appear in controlled combinations and then tested on molecules that contain unseen combinations of the same fragments. The training sets are deliberately chosen so that any model capturing the underlying physical rules of fragment interaction should be able to predict forces and energies accurately on the test molecules. Empirical evaluation demonstrates that errors on these out-of-distribution examples are typically an order of magnitude larger than on in-distribution examples, even when the models have been pre-trained on millions of other分子.
What carries the argument
A benchmark of four tasks that isolate compositional generalisation by holding out specific fragment combinations while ensuring the training distribution covers the physical principles needed to predict their interactions.
If this is right
- Current models primarily learn patterns tied to the specific training molecules rather than the compositional rules that determine molecular properties.
- Pre-training on millions of molecules does not close the generalization gap for these compositional tasks.
- Predictions for previously unseen molecules in applications such as drug design or materials discovery will carry substantially higher uncertainty.
- New model architectures or training strategies that explicitly encode fragment composition and physical combination rules are required to improve reliability.
Where Pith is reading between the lines
- The benchmark could be extended to other molecular properties such as electronic structure or reactivity to test whether the same compositional gap appears.
- If the performance gap persists across different model families it suggests that purely data-driven scaling may not be sufficient and hybrid physics-informed architectures may be necessary.
- Similar compositional benchmarks could be developed for related domains such as protein-ligand binding or crystal structure prediction to assess generalization limits more broadly.
Load-bearing premise
The training data is chosen such that generalisation to the test examples should be feasible for models that learn the underlying physical principles.
What would settle it
A single state-of-the-art model that achieves force and energy errors on the out-of-distribution test sets that are within a factor of two of its in-distribution errors would falsify the claim that the tasks are highly challenging for compositional generalisation.
Figures





















read the original abstract
Machine Learning Interatomic Potentials play a fundamental role in computational chemistry and materials science, enabling applications from molecular dynamics simulations to drug design and materials discovery. While recent approaches can estimate inter-atomic forces with high precision, it remains unclear to what extent they can generalise to previously unseen molecules. Do they learn the compositional structure of chemistry, capturing how molecular fragments and their combinations determine properties, or do they primarily learn to interpolate patterns that are specific to the training examples? To address this question, we propose a benchmark consisting of four tasks that require some form of compositional generalisation. In each task, models are tested on molecules that were unseen during training, but the training data is chosen such that generalisation to the test examples should be feasible for models that learn the underlying physical principles. Our empirical analysis shows that the considered tasks are highly challenging for state-of-the-art models, with errors on out-of-distribution examples often an order of magnitude higher than on in-distribution examples, even when using foundation models that have been pre-trained on millions of molecules.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a benchmark of four tasks to evaluate whether machine learning interatomic potentials (MLIPs) learn compositional structure in chemistry or merely interpolate training patterns. The central empirical claim is that state-of-the-art models, including foundation models pre-trained on millions of molecules, exhibit out-of-distribution (OOD) errors that are often an order of magnitude higher than in-distribution (ID) errors on these tasks, where the training data is asserted to allow generalization via underlying physical principles.
Significance. If the tasks are verifiably solvable by models that correctly extract physical composition rules, the benchmark would be a useful contribution for diagnosing generalization failures in MLIPs and guiding development of more principle-based models. The work is empirical and provides direct performance measurements without circular derivations.
major comments (2)
- [Abstract and task-construction sections] Abstract and task-construction sections: The claim that 'the training data is chosen such that generalisation to the test examples should be feasible for models that learn the underlying physical principles' is asserted without supporting evidence such as an oracle physics-based baseline (e.g., fixed force-field or fragment-additive model), formal argument that every test-molecule property is inferable from training atom-type/context combinations via known physical rules, or verification that no non-local effects or missing interaction types are required. This assumption is load-bearing for attributing the observed OOD/ID gap specifically to compositional-generalization failure rather than inherent task infeasibility.
- [Empirical-analysis section] Empirical-analysis section: The reported 'order of magnitude higher' OOD errors lack accompanying details on exact metrics, error-bar reporting, statistical controls, and explicit checks that the four tasks permit physical-principle-based generalization; without these, the central claim remains vulnerable to post-hoc task-design artifacts.
minor comments (2)
- [Figures and notation] Clarify notation for ID/OOD splits and ensure all figures include explicit legends distinguishing the four tasks and reporting both mean errors and variability.
- [Related work] Add a short related-work paragraph contrasting the proposed tasks with existing molecular generalization benchmarks to better situate the contribution.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We address each major comment below and have revised the manuscript accordingly to strengthen the justification of the benchmark and the reporting of results.
read point-by-point responses
-
Referee: [Abstract and task-construction sections] Abstract and task-construction sections: The claim that 'the training data is chosen such that generalisation to the test examples should be feasible for models that learn the underlying physical principles' is asserted without supporting evidence such as an oracle physics-based baseline (e.g., fixed force-field or fragment-additive model), formal argument that every test-molecule property is inferable from training atom-type/context combinations via known physical rules, or verification that no non-local effects or missing interaction types are required. This assumption is load-bearing for attributing the observed OOD/ID gap specifically to compositional-generalization failure rather than inherent task infeasibility.
Authors: We agree that the load-bearing assumption requires more explicit support. In the revised manuscript we have added, for each of the four tasks, a concise argument grounded in standard chemical principles (e.g., fragment additivity for energies and local-environment dependence for forces) together with a simple oracle baseline that implements a fragment-additive model using only quantities observable in the training set. This baseline recovers low error on the OOD test sets, indicating that the tasks are solvable once the relevant compositional rules are known. We also note that the tasks were deliberately constructed to avoid non-local or many-body effects outside the scope of the training atom-type and context combinations; any remaining higher-order interactions are negligible for the properties and molecular sizes considered. revision: yes
-
Referee: [Empirical-analysis section] Empirical-analysis section: The reported 'order of magnitude higher' OOD errors lack accompanying details on exact metrics, error-bar reporting, statistical controls, and explicit checks that the four tasks permit physical-principle-based generalization; without these, the central claim remains vulnerable to post-hoc task-design artifacts.
Authors: We have expanded the empirical-analysis section with the precise definitions of all reported metrics (MAE on energies and forces, with units), standard deviations computed over five independent training runs shown as error bars, and two-sided t-tests confirming that the OOD–ID gaps are statistically significant at p < 0.01 for every model and task. In addition, we now include a short verification subsection that cross-references each task to the physical principle it tests and confirms that the oracle baseline (described above) succeeds, thereby ruling out task infeasibility as the source of the observed gap. revision: yes
Circularity Check
No circularity: empirical benchmark with direct measurements
full rationale
This paper is a purely empirical benchmark study proposing four compositional generalization tasks for ML interatomic potentials and reporting measured error gaps between in-distribution and out-of-distribution examples. No derivations, equations, fitted parameters, or predictions appear in the provided text. The design statement that 'the training data is chosen such that generalisation to the test examples should be feasible for models that learn the underlying physical principles' is a methodological premise about task construction, not a quantity derived from or equivalent to the reported performance results. No self-citations, uniqueness theorems, or ansatzes are invoked in a load-bearing way. The central findings are direct empirical observations rather than quantities that reduce to the inputs by construction, satisfying the criteria for a self-contained benchmark without circularity.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
the training data is chosen such that generalisation to the test examples should be feasible for models that learn the underlying physical principles
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Our empirical analysis shows that the considered tasks are highly challenging for state-of-the-art models, with errors on out-of-distribution examples often an order of magnitude higher than on in-distribution examples
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
B. J. Alder and T. E. Wainwright. Studies in molecular dynamics. i. general method.The Journal of Chemical Physics, 31(2):459–466, 1959
work page 1959
- [2]
-
[3]
C. Bannwarth, S. Ehlert, and S. Grimme. Gfn2-xtb—an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density- dependent dispersion contributions.Journal of chemical theory and computation, 15(3):1652– 1671, 2019
work page 2019
-
[4]
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Csányi. Mace: Higher order equivari- ant message passing neural networks for fast and accurate force fields.Advances in neural information processing systems, 35:11423–11436, 2022
work page 2022
-
[5]
I. Batatia, P. Benner, Y . Chiang, A. M. Elena, D. P. Kovács, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, et al. A foundation model for atomistic materials chemistry. The Journal of chemical physics, 163(18), 2025
work page 2025
-
[6]
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials.Nature communications, 13(1):2453, 2022
work page 2022
- [7]
-
[8]
S. Chmiela, A. Tkatchenko, H. E. Sauceda, I. Poltavsky, K. T. Schütt, and K.-R. Müller. Machine learning of accurate energy-conserving molecular force fields.Science advances, 3(5):e1603015, 2017
work page 2017
-
[9]
S. Chmiela, V . Vassilev-Galindo, O. T. Unke, A. Kabylda, H. E. Sauceda, A. Tkatchenko, and K.-R. Müller. Accurate global machine learning force fields for molecules with hundreds of atoms.Science Advances, 9(2):eadf0873, 2023
work page 2023
-
[10]
B. Deng, Y . Choi, P. Zhong, J. Riebesell, S. Anand, Z. Li, K. Jun, K. A. Persson, and G. Ceder. Systematic softening in universal machine learning interatomic potentials.npj Computational Materials, 11(1):9, 2025
work page 2025
-
[11]
Y . Ektefaie, A. Shen, D. Bykova, M. G. Marin, M. Zitnik, and M. Farhat. Evaluating generaliz- ability of artificial intelligence models for molecular datasets.Nature Machine Intelligence, 6 (12):1512–1524, 2024. 10
work page 2024
-
[12]
D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke, and H. Jhoti. Twenty years on: the impact of fragments on drug discovery.Nature reviews Drug discovery, 15(9):605–619, 2016
work page 2016
-
[13]
R. Feng, Q. Zhu, H. Tran, B. Chen, A. Toland, R. Ramprasad, and C. Zhang. May the force be with you: Unified force-centric pre-training for 3d molecular conformations.Advances in neural information processing systems, 36:72750–72760, 2023
work page 2023
-
[14]
B. Focassio, L. P. M. Freitas, and G. R. Schleder. Performance assessment of universal machine learning interatomic potentials: Challenges and directions for materials’ surfaces.ACS Applied Materials & Interfaces, 17(9):13111–13121, 2024
work page 2024
-
[15]
J. A. Fodor and Z. W. Pylyshyn. Connectionism and cognitive architecture: A critical analysis. Cognition, 28(1-2):3–71, 1988
work page 1988
-
[16]
Margraf, and Stephan Günnemann
J. Gasteiger, S. Giri, J. T. Margraf, and S. Günnemann. Fast and uncertainty-aware directional message passing for non-equilibrium molecules.arXiv preprint arXiv:2011.14115, 2020
-
[17]
J. Gasteiger, F. Becker, and S. Günnemann. Gemnet: Universal directional graph neural networks for molecules.Advances in Neural Information Processing Systems, 34:6790–6802, 2021
work page 2021
-
[18]
Lawrence Zitnick, and Abhishek Das
J. Gasteiger, M. Shuaibi, A. Sriram, S. Günnemann, Z. Ulissi, C. L. Zitnick, and A. Das. Gemnet-oc: developing graph neural networks for large and diverse molecular simulation datasets.arXiv preprint arXiv:2204.02782, 2022
- [19]
-
[20]
D. Hupkes, V . Dankers, M. Mul, and E. Bruni. Compositionality decomposed: How do neural networks generalise?J. Artif. Intell. Res., 67:757–795, 2020. doi: 10.1613/JAIR.1.11674. URL https://doi.org/10.1613/jair.1.11674
-
[21]
Y . Ji, L. Zhang, J. Wu, B. Wu, L. Li, L.-K. Huang, T. Xu, Y . Rong, J. Ren, D. Xue, et al. Drugood: Out-of-distribution dataset curator and benchmark for ai-aided drug discovery–a focus on affinity prediction problems with noise annotations. InProceedings of the AAAI Conference on Artificial Intelligence, volume 37, pages 8023–8031, 2023
work page 2023
-
[22]
I. Khalid and S. Schockaert. Systematic relational reasoning with epistemic graph neural networks. InThe Thirteenth International Conference on Learning Representations, ICLR 2025, Singapore, April 24-28, 2025. OpenReview.net, 2025. URL https://openreview. net/forum?id=qNp86ByQlN
work page 2025
-
[23]
A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, et al. The atomic simulation environment—a python library for working with atoms.Journal of Physics: Condensed Matter, 29(27):273002, 2017
work page 2017
-
[24]
H. Li, X. Wang, Z. Zhang, and W. Zhu. Ood-gnn: Out-of-distribution generalized graph neural network.IEEE Transactions on Knowledge and Data Engineering, 35(7):7328–7340, 2022
work page 2022
- [25]
-
[26]
A. Mazitov, F. Bigi, M. Kellner, P. Pegolo, D. Tisi, G. Fraux, S. Pozdnyakov, P. Loche, and M. Ceriotti. Pet-mad as a lightweight universal interatomic potential for advanced materials modeling.Nature Communications, 16(1):10653, 2025
work page 2025
-
[27]
S. McIntosh-Smith, S. Alam, and C. Woods. Isambard-ai: a leadership-class supercomputer optimised specifically for artificial intelligence. InProceedings of the Cray User Group, pages 44–54. 2024
work page 2024
-
[28]
A. T. Muller, J. A. Hiss, and G. Schneider. Recurrent neural network model for constructive peptide design.Journal of chemical information and modeling, 58(2):472–479, 2018. 11
work page 2018
-
[29]
F. Neese. The orca program system.WIRES Comput. Molec. Sci., 2(1):73–78, 2012. doi: 10.1002/wcms.81
-
[30]
M. Neumann, J. Gin, B. Rhodes, S. Bennett, Z. Li, H. Choubisa, A. Hussey, and J. Godwin. Orb: A fast, scalable neural network potential.arXiv preprint arXiv:2410.22570, 2024
-
[31]
S. S. Omee, N. Fu, R. Dong, M. Hu, and J. Hu. Structure-based out-of-distribution (ood) materials property prediction: a benchmark study.npj Computational Materials, 10(1):144, 2024
work page 2024
-
[32]
S. Passaro and C. L. Zitnick. Reducing so (3) convolutions to so (2) for efficient equivariant gnns. InInternational conference on machine learning, pages 27420–27438. PMLR, 2023
work page 2023
-
[33]
Z. Pengmei, J. Liu, and Y . Shu. Beyond md17: the reactive xxmd dataset.Scientific Data, 11 (1):222, 2024
work page 2024
-
[34]
M. Pinheiro Jr, S. Zhang, P. O. Dral, and M. Barbatti. Ws22 database, wigner sampling and geometry interpolation for configurationally diverse molecular datasets.Scientific Data, 10(1): 95, 2023
work page 2023
-
[35]
A. Rahman. Correlations in the motion of atoms in liquid argon.Physical review, 136(2A): A405, 1964
work page 1964
-
[36]
R. Ramakrishnan, P. O. Dral, M. Rupp, and O. A. V on Lilienfeld. Quantum chemistry structures and properties of 134 kilo molecules.Scientific data, 1(1):1–7, 2014
work page 2014
-
[37]
Schlick.Molecular modeling and simulation: an interdisciplinary guide, volume 2
T. Schlick.Molecular modeling and simulation: an interdisciplinary guide, volume 2. Springer, 2010
work page 2010
-
[38]
M. Schreiner, A. Bhowmik, T. Vegge, J. Busk, and O. Winther. Transition1x-a dataset for building generalizable reactive machine learning potentials.Scientific Data, 9(1):779, 2022
work page 2022
- [39]
-
[40]
K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko, and K.-R. Müller. Schnet–a deep learning architecture for molecules and materials.The Journal of Chemical Physics, 148(24), 2018
work page 2018
-
[41]
J. S. Smith, O. Isayev, and A. E. Roitberg. Ani-1, a data set of 20 million calculated off- equilibrium conformations for organic molecules.Scientific data, 4(1):1–8, 2017
work page 2017
- [42]
-
[43]
K. Xu, J. Li, M. Zhang, S. S. Du, K. Kawarabayashi, and S. Jegelka. What can neural networks reason about? In8th International Conference on Learning Representations, ICLR 2020, Addis Ababa, Ethiopia, April 26-30, 2020. OpenReview.net, 2020. URL https://openreview. net/forum?id=rJxbJeHFPS
work page 2020
-
[44]
H. Zhou, A. Bradley, E. Littwin, N. Razin, O. Saremi, J. M. Susskind, S. Bengio, and P. Nakkiran. What algorithms can transformers learn? A study in length generalization. InThe Twelfth International Conference on Learning Representations, ICLR 2024, Vienna, Austria, May 7-11,
work page 2024
-
[45]
carboxylic acid is to alcohol+aldehyde as amide is to amine+aldehyde
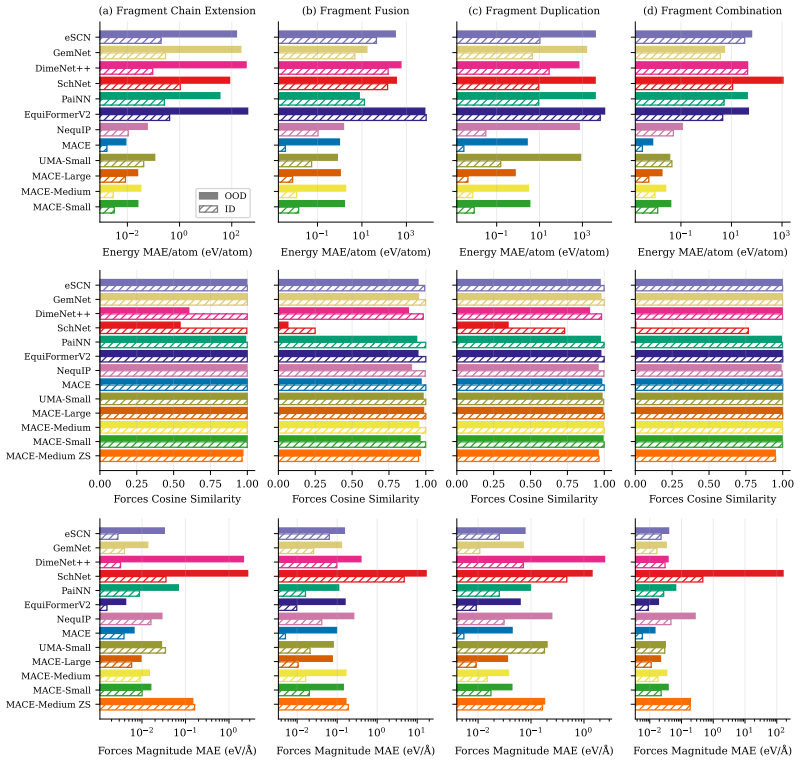
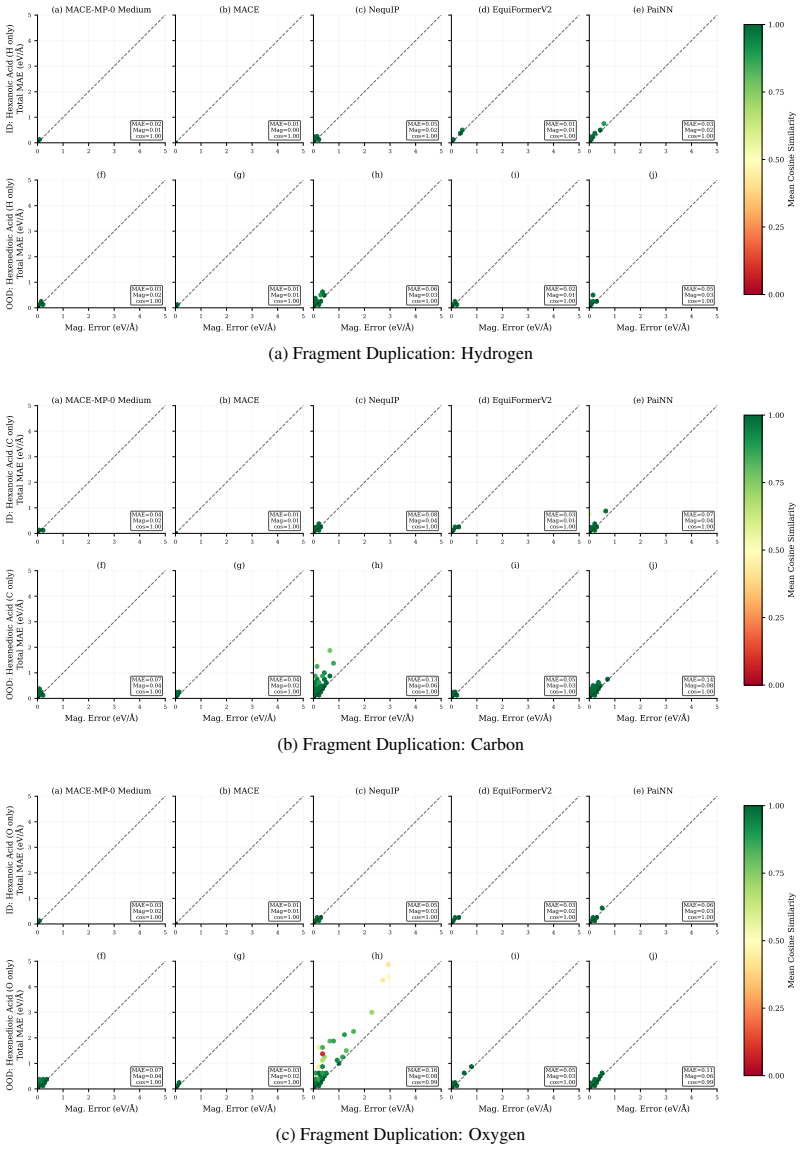
OpenReview.net, 2024. URLhttps://openreview.net/forum?id=AssIuHnmHX. 12 A Additional details and analysis A.1 Experiments with additional evaluation metrics Force MAE conflates errors in both the direction and magnitude of the predicted force vectors, while total energy MAE is inherently confounded by molecular size. To disentangle these distinct sources ...
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.