Recognition: no theorem link
Set-Aggregated Genome Embeddings for Microbiome Abundance Prediction
Pith reviewed 2026-05-13 02:56 UTC · model grok-4.3
The pith
Aggregating embeddings from genomic language models allows prediction of microbiome abundances and improves generalization to novel genomes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
In this work, we employ set-aggregated genome embeddings (SAGE) to predict community-level abundance profiles, exploiting the few-shot learning capabilities of genomic language models (GLMs). We benchmark this approach to show improved generalization on novel genomes compared to classical bioinformatics approaches. Model ablation shows that community-level latent representations directly result in improved performance. Lastly, we demonstrate the benefits of intermediate transformations between latent representations and demonstrate the differences between GLM embedding choices.
What carries the argument
Set-aggregated genome embeddings (SAGE), which pool individual genome embeddings produced by genomic language models to form a single community representation used for abundance prediction.
If this is right
- If correct, abundance profiles of microbial communities can be forecasted even when some genomes are new to the model.
- Community-level aggregation of embeddings yields better results than processing genomes separately.
- Applying intermediate transformations to the latent representations enhances predictive accuracy.
- The specific genomic language model chosen affects the quality of the resulting embeddings for this task.
Where Pith is reading between the lines
- The approach might apply to forecasting other community properties such as functional outputs or stability.
- It implies that genomic language models encode transferable functional information across different microbial species.
- Future work could test the method on real-world metagenomic samples with unknown compositions.
Load-bearing premise
That the embeddings generated by genomic language models contain sufficient functional information about genomes to enable accurate abundance prediction when aggregated, particularly for genomes not present in the training set.
What would settle it
A direct comparison of prediction error rates on a dataset of microbial communities where all test genomes are entirely absent from the training data; superior performance over classical methods would support the claim, while equivalent or worse performance would refute it.
Figures

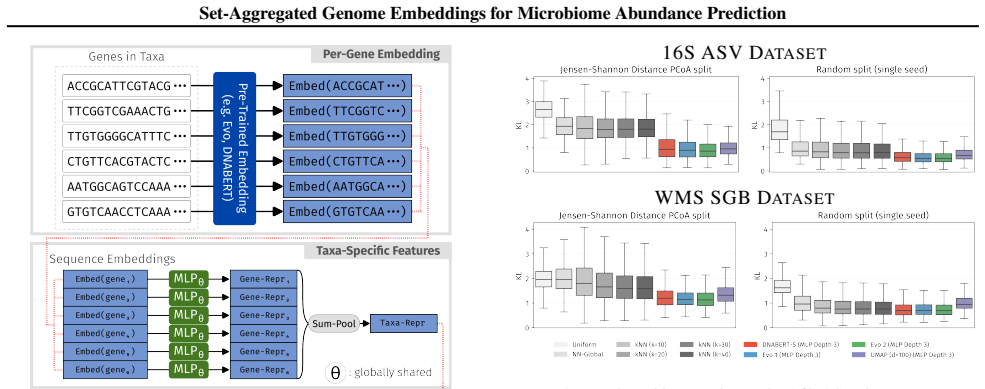

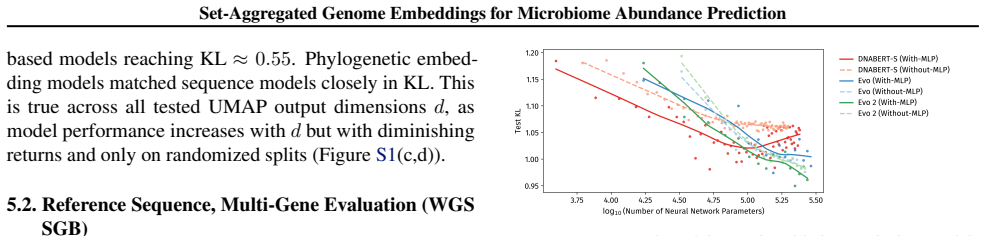

read the original abstract
Microbiome functions are encoded within the genes of the community-wide metagenome. A natural question is whether properties of a microbial community can be predicted just from knowing the raw DNA sequences of its members. In this work, we employ set-aggregated genome embeddings (SAGE) to predict community-level abundance profiles, exploiting the few-shot learning capabilities of genomic language models (GLMs). We benchmark this approach to show improved generalization on novel genomes compared to classical bioinformatics approaches. Model ablation shows that community-level latent representations directly result in improved performance. Lastly, we demonstrate the benefits of intermediate transformations between latent representations and demonstrate the differences between GLM embedding choices.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes set-aggregated genome embeddings (SAGE) from genomic language models (GLMs) to predict community-level microbiome abundance profiles directly from raw DNA sequences of community members. It benchmarks this against classical bioinformatics methods to claim improved generalization on novel genomes, with ablations demonstrating benefits of community-level latent representations and intermediate transformations between embeddings and predictions.
Significance. If the generalization claim holds under strict no-leakage conditions, the work would offer a valuable few-shot approach for metagenomic prediction by leveraging pre-trained GLM embeddings at the set level, potentially reducing the need for exhaustive training data in microbiome modeling. The ablations on latent representations and transformation choices provide useful empirical grounding for the method.
major comments (2)
- [Abstract and Methods] Abstract and Methods: The central generalization claim requires that 'novel genomes' in test communities are entirely absent from all training communities. No details are given on the genome-level partitioning procedure, whether splits are performed at the genome, sample, or read level, or on average sequence similarity between train and test genomes. This is load-bearing, as overlap would allow the set-aggregation model to exploit identity rather than functional embedding similarity, undermining attribution of gains to the GLM approach.
- [Results] Results (benchmarks and ablations): The reported improvements lack accompanying statistical tests, error bars, or details on data splits and cross-validation strategy. Without these, it is not possible to determine whether the performance differences versus classical baselines are robust or could be explained by partial genome overlap or split leakage.
minor comments (2)
- [Abstract and Introduction] The abstract and introduction would benefit from a clearer definition of 'set-aggregated' and how the aggregation operation is implemented (e.g., mean, attention, or other pooling) before the benchmarking claims.
- [Methods] Notation for the SAGE model and the intermediate transformations could be introduced more explicitly with a small diagram or pseudocode to aid reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments. We address each major comment point by point below and have revised the manuscript to provide the requested clarifications and statistical details.
read point-by-point responses
-
Referee: [Abstract and Methods] Abstract and Methods: The central generalization claim requires that 'novel genomes' in test communities are entirely absent from all training communities. No details are given on the genome-level partitioning procedure, whether splits are performed at the genome, sample, or read level, or on average sequence similarity between train and test genomes. This is load-bearing, as overlap would allow the set-aggregation model to exploit identity rather than functional embedding similarity, undermining attribution of gains to the GLM approach.
Authors: We agree that explicit documentation of the partitioning strategy is necessary to support the generalization claims. In the revised manuscript, we have expanded the Methods section with a full description of the genome-level partitioning procedure. Splits were performed at the genome level such that every genome appearing in any test community is entirely absent from all training communities. We have also added the average sequence similarity between train and test genomes, which is low enough to indicate that performance gains derive from functional embedding similarity rather than sequence identity. These additions directly address the concern about potential leakage. revision: yes
-
Referee: [Results] Results (benchmarks and ablations): The reported improvements lack accompanying statistical tests, error bars, or details on data splits and cross-validation strategy. Without these, it is not possible to determine whether the performance differences versus classical baselines are robust or could be explained by partial genome overlap or split leakage.
Authors: We acknowledge that statistical tests, error bars, and explicit split details are required for rigorous evaluation. The revised manuscript now includes paired statistical tests (with p-values) comparing SAGE against the classical baselines, error bars on all benchmark figures representing standard deviation across cross-validation folds, and a complete description of the data-splitting and cross-validation protocol in the Methods section. These changes allow readers to assess the robustness of the reported improvements independently of any potential overlap issues. revision: yes
Circularity Check
No significant circularity in derivation or claims
full rationale
The paper presents an empirical ML method that aggregates pre-trained genomic language model embeddings at the community level to predict abundances, with benchmarking against classical bioinformatics baselines and ablations on latent representations. No equations, derivations, or predictions are defined in terms of the target result itself. No self-citation chains, uniqueness theorems, or ansatzes are load-bearing for the central generalization claim. The approach is self-contained against external benchmarks and does not reduce any result to its inputs by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Genomic language models produce embeddings that encode functional properties relevant to community-level abundance.
Reference graph
Works this paper leans on
-
[1]
Nature Reviews Microbiology , volume=
Gut microbiota in human metabolic health and disease , author=. Nature Reviews Microbiology , volume=. 2021 , publisher=
work page 2021
-
[2]
Current understanding of the human microbiome , author=. Nature medicine , volume=. 2018 , publisher=
work page 2018
-
[3]
Nature Reviews Microbiology , volume=
Microbiota-mediated colonization resistance: mechanisms and regulation , author=. Nature Reviews Microbiology , volume=. 2023 , publisher=
work page 2023
-
[4]
Interaction between microbiota and immunity in health and disease , author=. Cell research , volume=. 2020 , publisher=
work page 2020
-
[5]
Trends in biotechnology , volume=
Synthetic biology tools to engineer microbial communities for biotechnology , author=. Trends in biotechnology , volume=. 2019 , publisher=
work page 2019
-
[6]
Diversity, stability and resilience of the human gut microbiota , author=. Nature , volume=. 2012 , publisher=
work page 2012
-
[7]
Nature Reviews Microbiology , volume=
Culturing the human microbiota and culturomics , author=. Nature Reviews Microbiology , volume=. 2018 , publisher=
work page 2018
-
[8]
Integration of 168,000 samples reveals global patterns of the human gut microbiome , author=. Cell , volume=. 2025 , publisher=
work page 2025
-
[9]
Specialized metabolic functions of keystone taxa sustain soil microbiome stability , author=. Microbiome , volume=. 2021 , publisher=
work page 2021
-
[10]
Recovery of the gut microbiota after antibiotics depends on host diet, community context, and environmental reservoirs , author=. Cell host & microbe , volume=. 2019 , publisher=
work page 2019
-
[11]
Large-scale association analyses identify host factors influencing human gut microbiome composition , author=. Nature genetics , volume=. 2021 , publisher=
work page 2021
-
[12]
Nature biotechnology , volume=
Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4 , author=. Nature biotechnology , volume=. 2023 , publisher=
work page 2023
-
[13]
Sequence modeling and design from molecular to genome scale with Evo , author=. Science , volume=. 2024 , publisher=
work page 2024
-
[14]
arXiv preprint arXiv:2402.08777 , volume=
Dnabert-s: Learning species-aware dna embedding with genome foundation models , author=. arXiv preprint arXiv:2402.08777 , volume=
-
[15]
DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome , author=. Bioinformatics , volume=. 2021 , publisher=
work page 2021
-
[16]
arXiv preprint arXiv:2306.15006 , year=
Dnabert-2: Efficient foundation model and benchmark for multi-species genome , author=. arXiv preprint arXiv:2306.15006 , year=
-
[17]
AlphaGenome: advancing regulatory variant effect prediction with a unified DNA sequence model , author=. BioRxiv , pages=. 2025 , publisher=
work page 2025
-
[18]
Longitudinal profiling of low-abundance strains in microbiomes with ChronoStrain , author=. Nature microbiology , volume=. 2025 , publisher=
work page 2025
-
[19]
Learning ecosystem-scale dynamics from microbiome data with MDSINE2 , author=. Nature Microbiology , volume=. 2025 , publisher=
work page 2025
-
[20]
Proceedings of the National Academy of Sciences , volume=
Physics-constrained neural ordinary differential equation models to discover and predict microbial community dynamics , author=. Proceedings of the National Academy of Sciences , volume=. 2026 , publisher=
work page 2026
-
[21]
Model-free prediction of microbiome compositions , author=. Microbiome , volume=. 2024 , publisher=
work page 2024
-
[22]
Genome modeling and design across all domains of life with Evo 2 , author=. BioRxiv , pages=. 2025 , publisher=
work page 2025
-
[23]
Nature communications , volume=
Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0 , author=. Nature communications , volume=. 2020 , publisher=
work page 2020
-
[24]
The human microbiome project , author=. Nature , volume=. 2007 , publisher=
work page 2007
-
[25]
SetBERT: the deep learning platform for contextualized embeddings and explainable predictions from high-throughput sequencing , author=. Bioinformatics , volume=. 2025 , publisher=
work page 2025
-
[26]
arXiv preprint arXiv:2508.11075 , year=
Abundance-Aware Set Transformer for Microbiome Sample Embedding , author=. arXiv preprint arXiv:2508.11075 , year=
-
[27]
A data-driven modeling framework for mapping genotypes to synthetic microbial community functions , author=. bioRxiv , year=
-
[28]
Advances in neural information processing systems , volume=
Attention is all you need , author=. Advances in neural information processing systems , volume=
-
[29]
Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines , author=. Nature Methods , volume=. 2025 , publisher=
work page 2025
-
[30]
American gut: an open platform for citizen science microbiome research , author=. Msystems , volume=. 2018 , publisher=
work page 2018
-
[31]
Assessment of metagenomic assembly using simulated next generation sequencing data , author=. PloS one , volume=. 2012 , publisher=
work page 2012
-
[32]
International journal of computer vision , volume=
Incremental learning for robust visual tracking , author=. International journal of computer vision , volume=. 2008 , publisher=
work page 2008
-
[33]
Some distance properties of latent root and vector methods used in multivariate analysis , author=. Biometrika , volume=. 1966 , publisher=
work page 1966
-
[34]
UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction
Umap: Uniform manifold approximation and projection for dimension reduction , author=. arXiv preprint arXiv:1802.03426 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[35]
Journal of translational medicine , volume=
Unraveling metagenomics through long-read sequencing: a comprehensive review , author=. Journal of translational medicine , volume=. 2024 , publisher=
work page 2024
-
[36]
Universality of human microbial dynamics , author=. Nature , volume=. 2016 , publisher=
work page 2016
-
[37]
Virtual Cell Challenge: Toward a Turing test for the virtual cell , author=. Cell , volume=. 2025 , publisher=
work page 2025
-
[38]
Gut microbiome heritability is nearly universal but environmentally contingent , author=. Science , volume=. 2021 , publisher=
work page 2021
-
[39]
High-resolution metagenome assembly for modern long reads with myloasm , author=. bioRxiv , year=
-
[40]
Nature Biotechnology , volume=
High-quality metagenome assembly from long accurate reads with metaMDBG , author=. Nature Biotechnology , volume=. 2024 , publisher=
work page 2024
-
[41]
metaFlye: scalable long-read metagenome assembly using repeat graphs , author=. Nature methods , volume=. 2020 , publisher=
work page 2020
-
[42]
Applied and environmental microbiology , volume=
Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities , author=. Applied and environmental microbiology , volume=. 2009 , publisher=
work page 2009
-
[43]
DADA2: High-resolution sample inference from Illumina amplicon data , author=. Nature methods , volume=. 2016 , publisher=
work page 2016
-
[44]
Nucleic acids research , volume=
MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform , author=. Nucleic acids research , volume=. 2002 , publisher=
work page 2002
-
[45]
Kraken: ultrafast metagenomic sequence classification using exact alignments , author=. Genome biology , volume=. 2014 , publisher=
work page 2014
- [46]
-
[47]
Ecological monographs , volume=
An ordination of the upland forest communities of southern Wisconsin , author=. Ecological monographs , volume=. 1957 , publisher=
work page 1957
-
[48]
European conference on computer vision , pages=
Visualizing and understanding convolutional networks , author=. European conference on computer vision , pages=. 2014 , organization=
work page 2014
-
[49]
arXiv preprint arXiv:1901.08644 , year=
Ablation studies in artificial neural networks , author=. arXiv preprint arXiv:1901.08644 , year=
-
[50]
International conference on machine learning , pages=
Set transformer: A framework for attention-based permutation-invariant neural networks , author=. International conference on machine learning , pages=. 2019 , organization=
work page 2019
-
[51]
Scaling Laws for Neural Language Models
Scaling laws for neural language models , author=. arXiv preprint arXiv:2001.08361 , year=
work page internal anchor Pith review Pith/arXiv arXiv 2001
-
[52]
Training Compute-Optimal Large Language Models
Training compute-optimal large language models , author=. arXiv preprint arXiv:2203.15556 , volume=
work page internal anchor Pith review Pith/arXiv arXiv
-
[53]
Comparing genomes recovered from time-series metagenomes using long-and short-read sequencing technologies , author=. Microbiome , volume=. 2023 , publisher=
work page 2023
-
[54]
Adam: A Method for Stochastic Optimization
Adam: A method for stochastic optimization , author=. arXiv preprint arXiv:1412.6980 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[55]
Strain tracking reveals the determinants of bacterial engraftment in the human gut following fecal microbiota transplantation , author=. Cell host & microbe , volume=. 2018 , publisher=
work page 2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.