Protein Fold Classification at Scale: Benchmarking and Pretraining
Pith reviewed 2026-05-20 13:11 UTC · model grok-4.3
The pith
Masked Invariant Autoencoders with up to 90 percent masking outperform supervised methods for protein fold classification on a new large benchmark.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
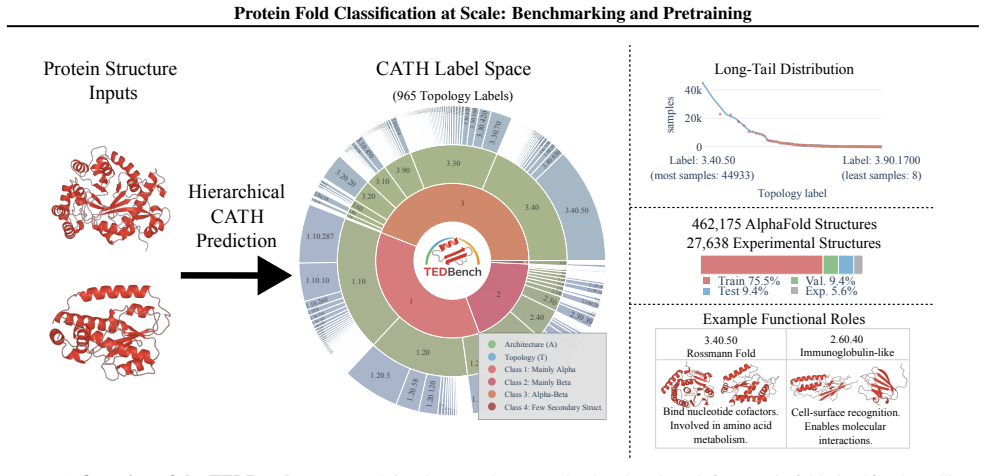
We introduce TEDBench, a large-scale, non-redundant benchmark for protein fold classification constructed from the Encyclopedia of Domains (TED) and Foldseek-clustered AlphaFold structures. We propose Masked Invariant Autoencoders (MiAE), a self-supervised framework for protein structure representation learning that uses an extremely high masking ratio of up to 90 percent with an SE(3)-invariant encoder and a lightweight decoder that reconstructs backbone coordinates from the latent representation and mask tokens. MiAE scales well and outperforms supervised counterparts and state-of-the-art baselines on TEDBench.
What carries the argument
Masked Invariant Autoencoders (MiAE) that apply extreme masking to protein backbone coordinates and use an SE(3)-invariant encoder with a lightweight decoder to reconstruct the full structure from the masked latent representation.
If this is right
- Protein fold classification benefits from self-supervised pretraining on large unlabeled structure sets instead of relying solely on supervised training.
- Very high masking ratios remain effective for learning useful structural representations in this domain.
- The non-redundant benchmark construction removes duplicate-induced inflation that affected earlier protein fold datasets.
- Gains from MiAE pretraining carry over when models are tested on experimental rather than predicted structures.
Where Pith is reading between the lines
- The same high-masking recipe might reduce the model size needed for other structure-related prediction tasks in biology.
- If AlphaFold models contain systematic local errors, future benchmarks could combine experimental and predicted data with explicit error weighting.
- Similar masking-based pretraining could be tested on other chain-like biomolecules such as RNA backbones.
Load-bearing premise
The construction of TEDBench from TED and Foldseek-clustered AlphaFold structures yields a truly non-redundant and unbiased test of fold classification that generalizes beyond prediction artifacts in AlphaFold models.
What would settle it
Re-evaluate all models on a version of the benchmark rebuilt by clustering only experimental CATH structures with an independent algorithm; if MiAE loses its performance advantage or if substantial fold duplicates appear in the test split, the central scaling claim would be falsified.
Figures





read the original abstract
Classifying protein topology is essential for deciphering biological function, but progress is held back by the lack of large-scale benchmarks that avoid duplicates and by models that do not scale well. We introduce TEDBench, a large-scale, non-redundant benchmark for protein fold classification constructed from the Encyclopedia of Domains (TED) and Foldseek-clustered AlphaFold structures. We show that on TEDBench, current protein representation learning methods either require very large models or fail to deliver strong performance. To address this challenge, we propose Masked Invariant Autoencoders (MiAE), a self-supervised framework for protein structure representation learning. MiAE uses an extremely high masking ratio of up to 90% with an $\mathrm{SE(3)}$-invariant encoder and a lightweight decoder that reconstructs backbone coordinates from the latent representation and mask tokens. MiAE scales well and outperforms supervised counterparts and state-of-the-art baselines on TEDBench, establishing a strong recipe for protein fold classification. To test transfer beyond AlphaFold structures, we further benchmark on a curated dataset from experimental structures of CATH v4.4. TEDBench is available at https://github.com/BorgwardtLab/TEDBench.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces TEDBench, a large-scale non-redundant benchmark for protein fold classification built from the Encyclopedia of Domains (TED) combined with Foldseek-clustered AlphaFold structures. It proposes Masked Invariant Autoencoders (MiAE), a self-supervised framework using up to 90% masking, an SE(3)-invariant encoder, and a lightweight decoder that reconstructs backbone coordinates. The central claim is that MiAE scales well and outperforms both supervised baselines and prior state-of-the-art methods on TEDBench while transferring to a curated experimental CATH v4.4 dataset; TEDBench is released publicly.
Significance. If the results hold after validation of the benchmark, the work would be significant for structural bioinformatics and representation learning by supplying a scalable self-supervised recipe for fold classification and a new public benchmark that addresses duplication issues in existing resources. The high-masking invariant autoencoder approach and the github release of TEDBench constitute concrete contributions to reproducibility.
major comments (2)
- [TEDBench construction] TEDBench construction section: the description of combining TED domains with Foldseek-clustered AlphaFold structures supplies no quantitative details on clustering thresholds, maximum inter-set TM-scores, sequence-identity cutoffs, or explicit checks that train/test folds are structurally dissimilar. Without these metrics it is impossible to confirm that reported gains reflect learned fold representations rather than residual similarities or AlphaFold geometric biases; this directly undermines the central outperformance claim.
- [Results] Results and experimental sections: the claim that MiAE 'outperforms supervised counterparts and state-of-the-art baselines' on TEDBench and transfers to CATH requires tabulated metrics with error bars, ablation studies on the masking ratio, and precise baseline implementations. The abstract states clear superiority but the manuscript must supply these numbers to substantiate scaling behavior and rule out dataset-specific artifacts.
minor comments (2)
- [Abstract] Abstract: the phrase 'up to 90%' masking ratio should be accompanied by the exact ratio(s) used in the reported experiments for clarity.
- [Methods] Notation: the SE(3)-invariant encoder is introduced without an explicit equation or reference to the invariance property; adding a short formal definition would improve readability.
Simulated Author's Rebuttal
We thank the referee for their thorough and constructive review of our manuscript. We have carefully considered the two major comments and provide point-by-point responses below. We agree that both points identify areas where the manuscript can be strengthened and plan to make the corresponding revisions.
read point-by-point responses
-
Referee: [TEDBench construction] TEDBench construction section: the description of combining TED domains with Foldseek-clustered AlphaFold structures supplies no quantitative details on clustering thresholds, maximum inter-set TM-scores, sequence-identity cutoffs, or explicit checks that train/test folds are structurally dissimilar. Without these metrics it is impossible to confirm that reported gains reflect learned fold representations rather than residual similarities or AlphaFold geometric biases; this directly undermines the central outperformance claim.
Authors: We agree that additional quantitative details on benchmark construction are required for full reproducibility and to allow independent verification that train/test splits are structurally dissimilar. In the revised manuscript we will expand the TEDBench construction section to report the specific Foldseek clustering parameters, maximum inter-set TM-scores, sequence-identity cutoffs, and the results of explicit structural dissimilarity checks (e.g., TM-align) between folds in different splits. These metrics will be presented in the main text together with a supplementary table summarizing the final dataset statistics and split properties. revision: yes
-
Referee: [Results] Results and experimental sections: the claim that MiAE 'outperforms supervised counterparts and state-of-the-art baselines' on TEDBench and transfers to CATH requires tabulated metrics with error bars, ablation studies on the masking ratio, and precise baseline implementations. The abstract states clear superiority but the manuscript must supply these numbers to substantiate scaling behavior and rule out dataset-specific artifacts.
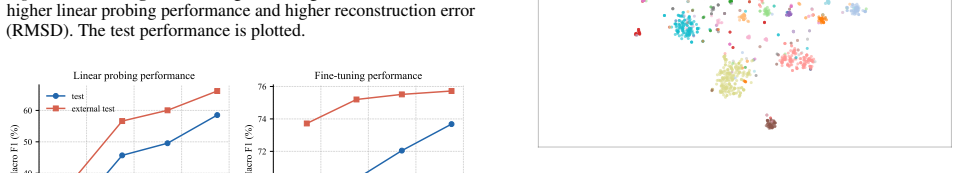
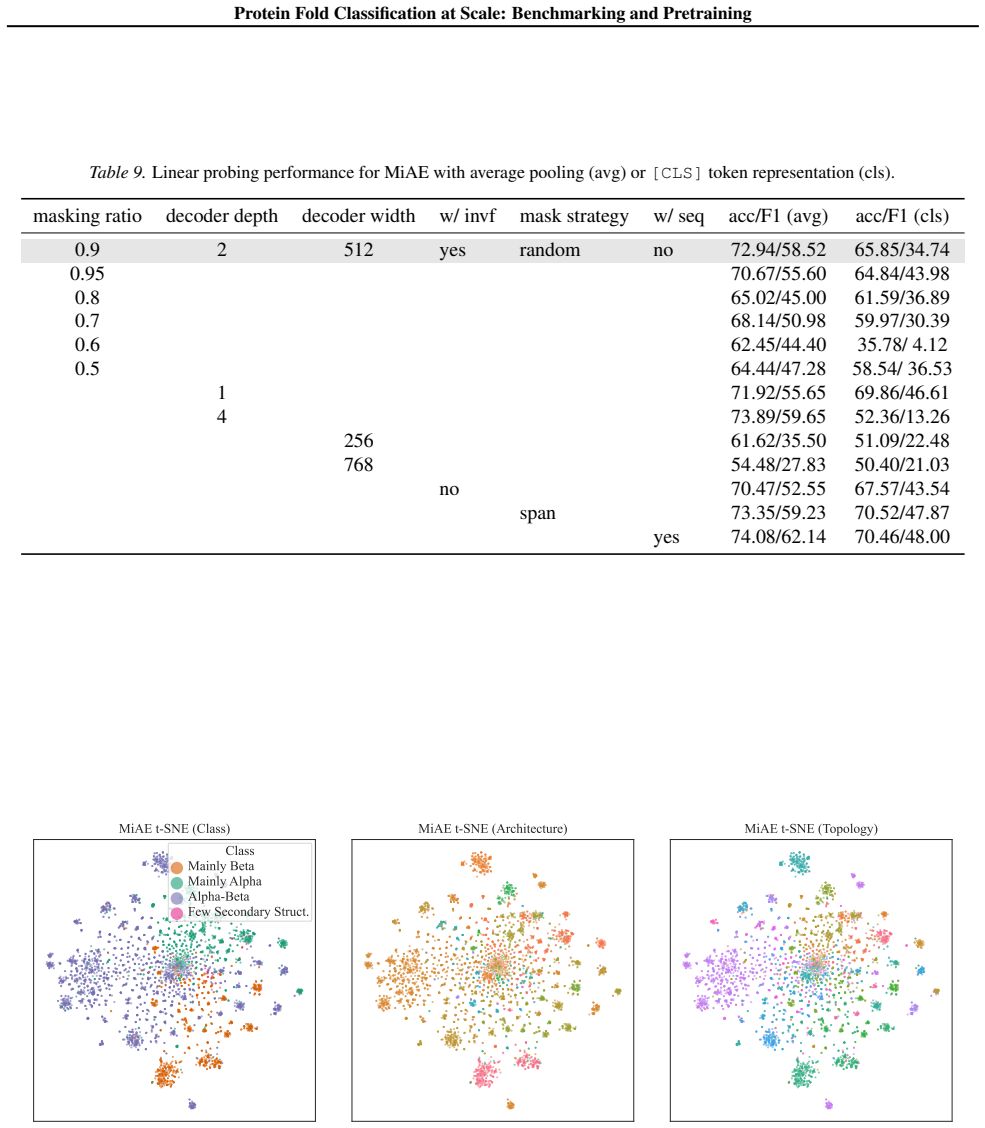
Authors: We acknowledge that the experimental reporting can be made more rigorous. In the revised manuscript we will augment the results section with complete tabulated performance metrics (including accuracy, F1, and other relevant measures) accompanied by error bars from multiple independent runs, dedicated ablation experiments varying the masking ratio, and precise descriptions of all baseline implementations (model architectures, hyperparameters, and training protocols). These additions will directly support the scaling claims and outperformance statements. revision: yes
Circularity Check
New benchmark construction and self-supervised pretraining exhibit no self-referential reduction or load-bearing self-citation.
full rationale
The paper constructs TEDBench from TED domains plus Foldseek-clustered AlphaFold structures and then evaluates MiAE (high-masking SE(3)-invariant autoencoder) on it, reporting empirical outperformance over supervised baselines and prior methods. No equations, fitted parameters renamed as predictions, or self-citation chains are visible that would make the reported gains equivalent to the inputs by construction. The CATH experimental benchmark is invoked as an external transfer test. This is the common honest case of a self-contained empirical contribution; the reader's assigned score of 2 reflects only the normal novelty of a new benchmark rather than any circular step.
Axiom & Free-Parameter Ledger
free parameters (1)
- masking ratio
axioms (1)
- domain assumption Protein backbone coordinates are SE(3)-invariant
invented entities (1)
-
MiAE
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Highly accurate protein structure prediction with AlphaFold , author=. nature , volume=. 2021 , publisher=
work page 2021
-
[2]
Nucleic acids research , volume=
AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences , author=. Nucleic acids research , volume=. 2024 , publisher=
work page 2024
-
[3]
Deng, Jia and Dong, Wei and Socher, Richard and Li, Li-Jia and Li, Kai and Fei-Fei, Li , booktitle=cvpr, pages=. 2009 , organization=
work page 2009
-
[4]
The shape and structure of proteins , author=. Molecular Biology of the Cell. 4th edition , year=
-
[5]
Computational and structural biotechnology journal , volume=
Protein domain identification methods and online resources , author=. Computational and structural biotechnology journal , volume=. 2021 , publisher=
work page 2021
-
[6]
Orengo, Christine A and Michie, Alex D and Jones, Susan and Jones, David T and Swindells, Mark B and Thornton, Janet M , journal=. 1997 , publisher=
work page 1997
-
[7]
Waman, Vaishali P and Bordin, Nicola and Lau, Andy and Kandathil, Shaun and Wells, Jude and Miller, David and Velankar, Sameer and Jones, David T and Sillitoe, Ian and Orengo, Christine , journal=. 2025 , publisher=
work page 2025
-
[8]
Redfern, Oliver C and Harrison, Andrew and Dallman, Tim and Pearl, Frances M G and Orengo, Christine A , journal=. 2007 , publisher=
work page 2007
-
[9]
Nallapareddy, Vamsi and Bordin, Nicola and Sillitoe, Ian and Heinzinger, Michael and Littmann, Maria and Waman, Vaishali P and Sen, Neeladri and Rost, Burkhard and Orengo, Christine , journal=. 2023 , publisher=
work page 2023
-
[10]
Dawson, Natalie L and Lewis, Tony E and Das, Sayoni and Lees, Jonathan G and Lee, David and Ashford, Paul and Orengo, Christine A and Sillitoe, Ian , journal=. 2017 , publisher=
work page 2017
-
[11]
Csaba, Gergely and Birzele, Fabian and Zimmer, Ralf , journal=. Systematic comparison of. 2009 , publisher=
work page 2009
-
[12]
Exploring structural diversity across the protein universe with The Encyclopedia of Domains , author=. Science , volume=. 2024 , publisher=
work page 2024
-
[13]
Nature biotechnology , volume=
Fast and accurate protein structure search with Foldseek , author=. Nature biotechnology , volume=. 2024 , publisher=
work page 2024
-
[14]
Nature Communications , volume=
Merizo: a rapid and accurate protein domain segmentation method using invariant point attention , author=. Nature Communications , volume=. 2023 , publisher=
work page 2023
-
[15]
Chainsaw: protein domain segmentation with fully convolutional neural networks , author=. Bioinformatics , volume=. 2024 , publisher=
work page 2024
-
[16]
A unified approach to protein domain parsing with inter-residue distance matrix , author=. Bioinformatics , volume=. 2023 , publisher=
work page 2023
-
[17]
Hou, Jie and Adhikari, Badri and Cheng, Jianlin , journal=. 2018 , publisher=
work page 2018
-
[18]
Clustering predicted structures at the scale of the known protein universe , author=. Nature , volume=. 2023 , publisher=
work page 2023
-
[19]
Masked autoencoders are scalable vision learners , author=
-
[20]
Simulating 500 million years of evolution with a language model , author=. Science , volume=. 2025 , publisher=
work page 2025
-
[21]
Generative models for graph-based protein design , author=
-
[22]
Roformer: Enhanced transformer with rotary position embedding , author=. Neurocomputing , volume=. 2024 , publisher=
work page 2024
-
[23]
Holm, L. and Sander, C. , title =. J. Mol. Biol. , volume =. 1993 , month = sep, issn =. 8377180 , doi =
work page 1993
-
[24]
and Harrison, Andrew and Dallman, Tim and Pearl, Frances M
Redfern, Oliver C. and Harrison, Andrew and Dallman, Tim and Pearl, Frances M. G. and Orengo, Christine A. , title =. PLoS Comput. Biol. , volume =. 2007 , month = nov, issn =. 18052539 , doi =
work page 2007
-
[25]
Shindyalov, I. N. and Bourne, P. E. , title =. Protein Eng. , volume =. 1998 , month = sep, issn =. 9796821 , doi =
work page 1998
-
[26]
UniProt: the uni- versal protein knowledgebase in 2025
The UniProt Consortium , title =. Nucleic Acids Research , volume =. 2024 , month =. doi:10.1093/nar/gkae1010 , url =
-
[27]
M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T
Berman, Helen M. and Westbrook, John and Feng, Zukang and Gilliland, Gary and Bhat, T. N. and Weissig, Helge and Shindyalov, Ilya N. and Bourne, Philip E. , title =. Nucleic Acids Research , volume =. 2000 , month =. doi:10.1093/nar/28.1.235 , url =
-
[28]
Geometric Deep Learning: Grids, Groups, Graphs, Geodesics, and Gauges
Bronstein, Michael M. and Bruna, Joan and Cohen, Taco and Veli. arXiv , year =. 2104.13478 , doi =
work page internal anchor Pith review Pith/arXiv arXiv
-
[29]
Mario Geiger and Tess Smidt and Alby M. and Benjamin Kurt Miller and Wouter Boomsma and Bradley Dice and Kostiantyn Lapchevskyi and Maurice Weiler and Michał Tyszkiewicz and Simon Batzner and Dylan Madisetti and Martin Uhrin and Jes Frellsen and Nuri Jung and Sophia Sanborn and Mingjian Wen and Josh Rackers and Marcel Rød and Michael Bailey , title =. doi...
-
[30]
Ilyes Batatia and David Peter Kovacs and Gregor N. C. Simm and Christoph Ortner and Gabor Csanyi , booktitle=. 2022 , url=
work page 2022
- [31]
-
[32]
J. Dauparas and I. Anishchenko and N. Bennett and H. Bai and R. J. Ragotte and L. F. Milles and B. I. M. Wicky and A. Courbet and R. J. de Haas and N. Bethel and P. J. Y. Leung and T. F. Huddy and S. Pellock and D. Tischer and F. Chan and B. Koepnick and H. Nguyen and A. Kang and B. Sankaran and A. K. Bera and N. P. King and D. Baker , title =. Science , ...
work page 2022
-
[33]
Gilmer, Justin and Schoenholz, Samuel S. and Riley, Patrick F. and Vinyals, Oriol and Dahl, George E. , title =. 2017 , publisher =
work page 2017
-
[34]
The Graph Neural Network Model , year=
Scarselli, Franco and Gori, Marco and Tsoi, Ah Chung and Hagenbuchner, Markus and Monfardini, Gabriele , journal=. The Graph Neural Network Model , year=
-
[35]
Protein Engineering, Design and Selection , volume =
Yang, Kevin K and Zanichelli, Niccolò and Yeh, Hugh , title =. Protein Engineering, Design and Selection , volume =. 2022 , month =. doi:10.1093/protein/gzad015 , url =
-
[36]
Attention is All you Need , url =
Vaswani, Ashish and Shazeer, Noam and Parmar, Niki and Uszkoreit, Jakob and Jones, Llion and Gomez, Aidan N and Kaiser, ukasz and Polosukhin, Illia , booktitle =. Attention is All you Need , url =
-
[37]
SaProt: Protein Language Modeling with Structure-aware Vocabulary , author=. 2024 , url=
work page 2024
-
[38]
Evolutionary-scale prediction of atomic-level protein structure with a language model , author=. Science , volume=. 2023 , publisher=
work page 2023
-
[39]
Bert: Pre-training of deep bidirectional transformers for language understanding , author=
-
[40]
Proceedings of the National Academy of Sciences , volume=
Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences , author=. Proceedings of the National Academy of Sciences , volume=. 2021 , publisher=
work page 2021
-
[41]
Endowing protein language models with structural knowledge , author=. Bioinformatics , volume=. 2025 , publisher=
work page 2025
-
[42]
Protein Representation Learning by Geometric Structure Pretraining , author=
-
[43]
Zhu, Ciyou and Byrd, Richard H. and Lu, Peihuang and Nocedal, Jorge , title =. ACM Trans. Math. Softw. , month = dec, pages =. 1997 , issue_date =. doi:10.1145/279232.279236 , abstract =
-
[44]
ELECTRA: Pre-training Text Encoders as Discriminators Rather Than Generators
Electra: Pre-training text encoders as discriminators rather than generators , author=. arXiv preprint arXiv:2003.10555 , year=
work page internal anchor Pith review Pith/arXiv arXiv 2003
-
[45]
Journal of Machine Learning Research , year =
Laurens van der Maaten and Geoffrey Hinton , title =. Journal of Machine Learning Research , year =
-
[46]
Keller Jordan and Yuchen Jin and Vlado Boza and You Jiacheng and Franz Cesista and Laker Newhouse and Jeremy Bernstein , title =. 2024 , url =
work page 2024
-
[47]
International Conference on Learning Representations , year=
Decoupled Weight Decay Regularization , author=. International Conference on Learning Representations , year=
-
[48]
Kingma and Jimmy Ba , editor =
Diederik P. Kingma and Jimmy Ba , editor =. Adam:. 3rd International Conference on Learning Representations,. 2015 , url =
work page 2015
-
[49]
Proceedings of the National Academy of Sciences , volume=
Tertiary alphabet for the observable protein structural universe , author=. Proceedings of the National Academy of Sciences , volume=. 2016 , publisher=
work page 2016
-
[50]
Proteinshake: Building datasets and benchmarks for deep learning on protein structures , author=
-
[51]
Evaluating protein transfer learning with TAPE , author=
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.