Atom-level Protein Representation Learning Improves Protein Structure Prediction
Pith reviewed 2026-05-25 02:52 UTC · model grok-4.3
The pith
Pretraining to recover tokens from corrupted amino-acid, backbone, and full-atom views produces representations that improve structure-prediction tasks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
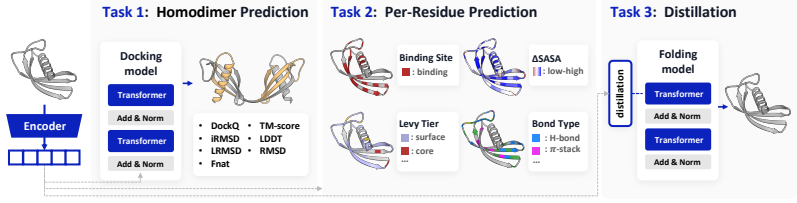
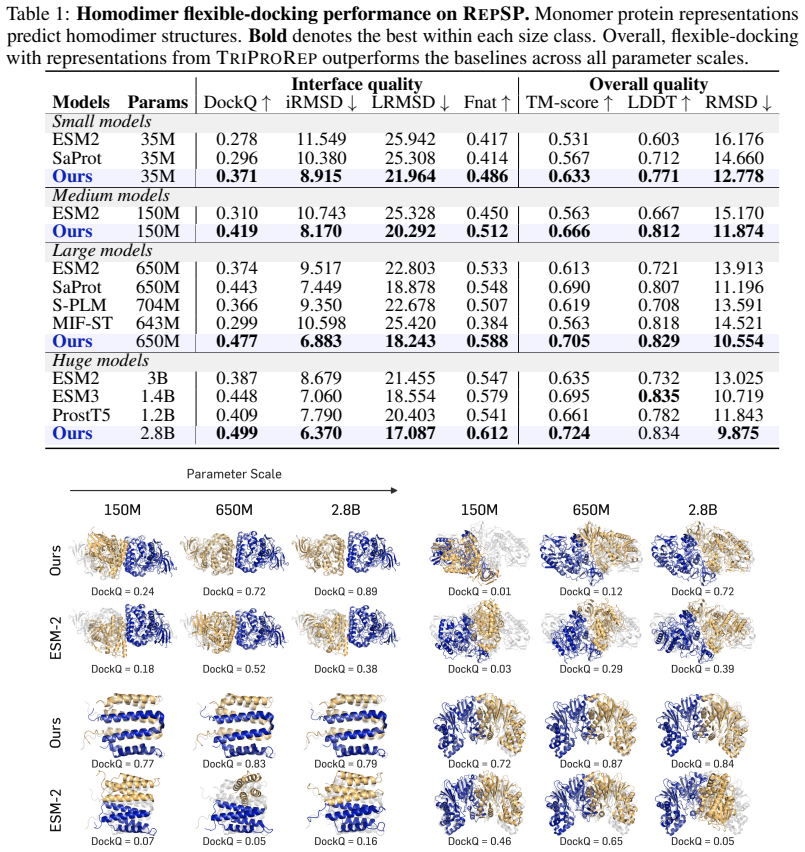
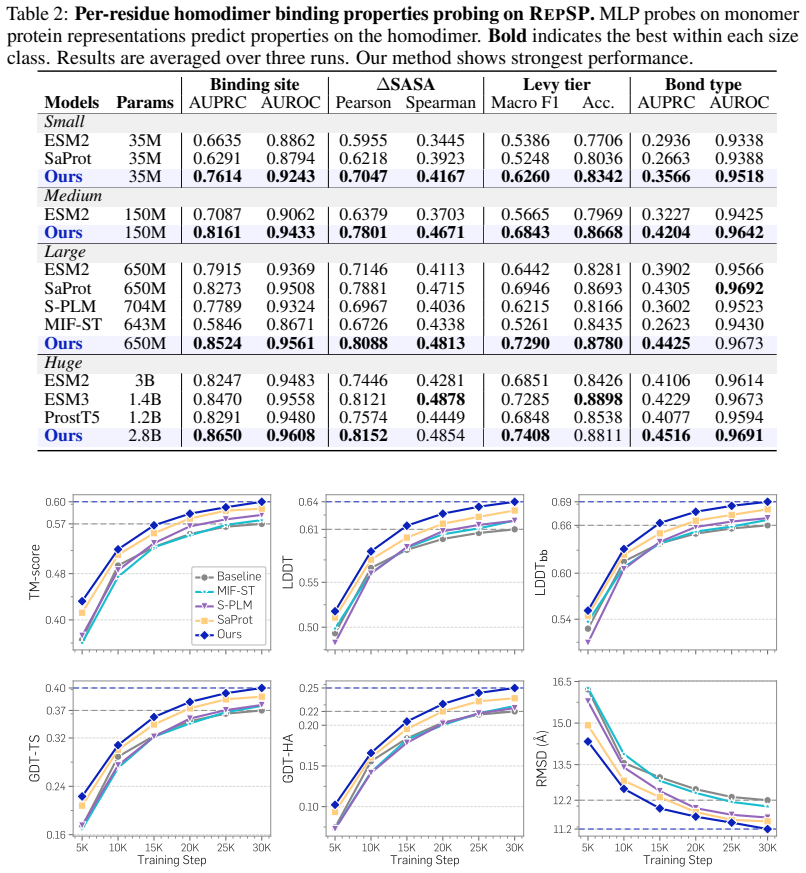
TriProRep jointly models three aligned residue-level views—amino-acid identity, backbone geometry, and local full-atom geometry—discretely encoded via VQ-VAE tokenizers. By pretraining to recover original tokens from generator-corrupted views, it learns to distinguish plausible but incorrect cross-view augmentations from the original protein. When evaluated on RepSP, which includes homodimer co-folding from apo-chain representations, residue-level prediction of interaction properties, and representation-aligned monomer structure prediction, TriProRep improves over sequence-only and prior structure-aware models.
What carries the argument
Joint three-view token recovery pretraining on VQ-VAE-encoded amino-acid identity, backbone geometry, and local full-atom geometry.
If this is right
- Representations support direct homodimer co-folding from individual apo-chain inputs.
- They improve residue-level prediction of homodimer interaction properties.
- They enhance monomer structure prediction when the model is aligned to the learned representations.
- Performance on conventional protein benchmarks remains competitive with prior methods.
Where Pith is reading between the lines
- The same cross-view corruption objective could be tested on other biomolecules that have both sequence and coordinate data.
- RepSP-style benchmarks may expose limitations in existing representations when the downstream goal is interaction rather than single-chain folding.
- If the three-view alignment proves robust, similar tokenization could reduce the need for task-specific fine-tuning in related prediction problems.
Load-bearing premise
Recovering original tokens from generator-corrupted cross-view augmentations produces representations that transfer to downstream structure-prediction tasks without additional fine-tuning or task-specific adaptation.
What would settle it
Apply the frozen TriProRep representations to the three RepSP tasks and measure whether co-folding accuracy, interaction prediction AUC, or monomer structure quality show no improvement over sequence-only baselines.
Figures


read the original abstract
Recent advances in generative modeling show that pretrained representations can improve generation as conditioning features or alignment targets. Motivated by this, we study protein representations for predicting structures beyond conventional function annotation. We propose TriProRep, a structure-aware pretraining method that jointly models three aligned residue-level views: amino-acid identity, backbone geometry, and local full-atom geometry, discretely encoded via VQ-VAE tokenizers. By pretraining to recover original tokens from generator-corrupted views, TriProRep learns to distinguish plausible but incorrect cross-view augmentations from the original protein. We further introduce RepSP, a benchmark for evaluating protein representations in structure-predictive settings. RepSP tests three uses of representations: homodimer co-folding from apo-chain representations, residue-level prediction of homodimer-derived interaction properties, and representation-aligned monomer structure prediction. Across these tasks, TriProRep improves over sequence-only and prior structure-aware representation models, while maintaining competitive performance on conventional benchmarks.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes TriProRep, a structure-aware pretraining method that jointly models three aligned residue-level views (amino-acid identity, backbone geometry, and local full-atom geometry) via VQ-VAE tokenizers. Pretraining recovers original tokens from generator-corrupted cross-view augmentations to learn distinctions between plausible but incorrect augmentations and the original protein. The authors introduce the RepSP benchmark, which evaluates representations on homodimer co-folding from apo-chain representations, residue-level prediction of homodimer-derived interaction properties, and representation-aligned monomer structure prediction. They claim TriProRep improves over sequence-only and prior structure-aware models on RepSP tasks while remaining competitive on conventional benchmarks.
Significance. If the claimed improvements hold under rigorous controls, the work would demonstrate that atom-level cross-view token recovery pretraining can yield transferable structure-aware representations without task-specific fine-tuning, advancing representation learning for proteins beyond sequence-only or coarse structure models. The RepSP benchmark itself would be a useful standardized testbed for structure-predictive uses of representations.
major comments (2)
- [Abstract] Abstract: the claim of improvements across RepSP tasks is stated without any quantitative results, baselines, error bars, ablation studies, or statistical details, preventing assessment of whether the gains are load-bearing or attributable to the proposed pretraining rather than dataset effects.
- [Abstract] Abstract (pretraining description): the token-recovery objective on generator-corrupted cross-view augmentations is presented as producing representations that improve homodimer co-folding, interaction prediction, and monomer folding without fine-tuning, yet the objective supplies only local cross-view consistency and no direct supervision on global fold geometry or interaction interfaces; this leaves open whether reported gains reflect learned structure awareness or artifacts such as tokenizer leakage or data overlap.
minor comments (1)
- [Abstract] Abstract: 'RepSP' is introduced as a new benchmark without spelling out the acronym or providing even a high-level description of its three tasks beyond the listed uses.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We address each major comment below and indicate where revisions will be made to strengthen the presentation.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim of improvements across RepSP tasks is stated without any quantitative results, baselines, error bars, ablation studies, or statistical details, preventing assessment of whether the gains are load-bearing or attributable to the proposed pretraining rather than dataset effects.
Authors: We agree that the abstract would benefit from quantitative support. In the revised manuscript, we will incorporate key performance metrics from the RepSP benchmark, including specific improvements over sequence-only and prior structure-aware baselines, along with error bars from repeated runs and any available statistical details. This will allow clearer evaluation of the gains. revision: yes
-
Referee: [Abstract] Abstract (pretraining description): the token-recovery objective on generator-corrupted cross-view augmentations is presented as producing representations that improve homodimer co-folding, interaction prediction, and monomer folding without fine-tuning, yet the objective supplies only local cross-view consistency and no direct supervision on global fold geometry or interaction interfaces; this leaves open whether reported gains reflect learned structure awareness or artifacts such as tokenizer leakage or data overlap.
Authors: While the objective operates at the local residue level, the joint three-view modeling is intended to capture structural distinctions that transfer to global tasks, as demonstrated by the RepSP results on co-folding and interaction properties. The full manuscript provides comparisons showing advantages over prior models. To address concerns about artifacts, we will expand the discussion section with additional analysis on data overlap and tokenizer behavior. We view the gains as reflecting the learned representations rather than artifacts. revision: partial
Circularity Check
No significant circularity: pretraining objective independent of downstream RepSP tasks
full rationale
The paper defines TriProRep via a VQ-VAE token-recovery objective on generator-corrupted cross-view augmentations (amino-acid, backbone, full-atom). This objective is stated independently of the RepSP benchmark tasks (homodimer co-folding, residue-level interaction prediction, representation-aligned monomer folding). No equations, fitted parameters, or self-citations are shown that reduce the claimed gains on RepSP to quantities fitted on the evaluation data or to self-referential definitions. The derivation chain remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption VQ-VAE tokenizers can faithfully discretize amino-acid identity, backbone geometry, and local full-atom geometry without significant information loss for downstream structure tasks.
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
pretraining to recover original tokens from generator-corrupted views... three aligned residue-level views
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
VQ-VAE tokenizers... corrective pretraining objective
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.