Benchmarking Pathology Foundation Models for Spatial Domain Understanding
Pith reviewed 2026-06-29 22:45 UTC · model grok-4.3
The pith
Pathology foundation models capture distinct aspects of tissue spatial architecture depending on pretraining method.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
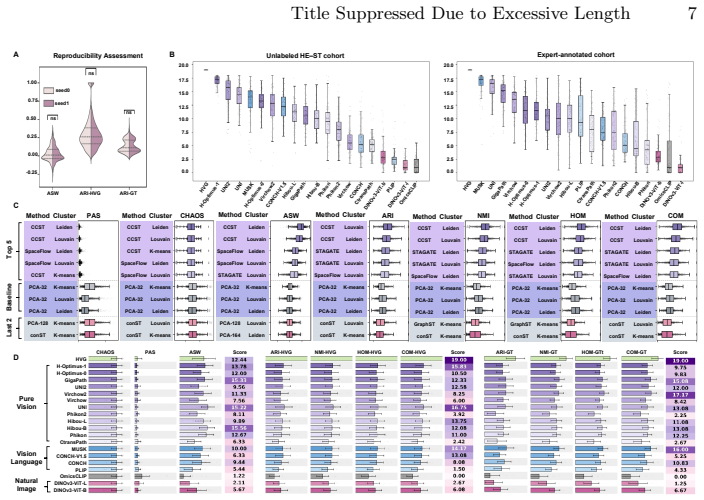
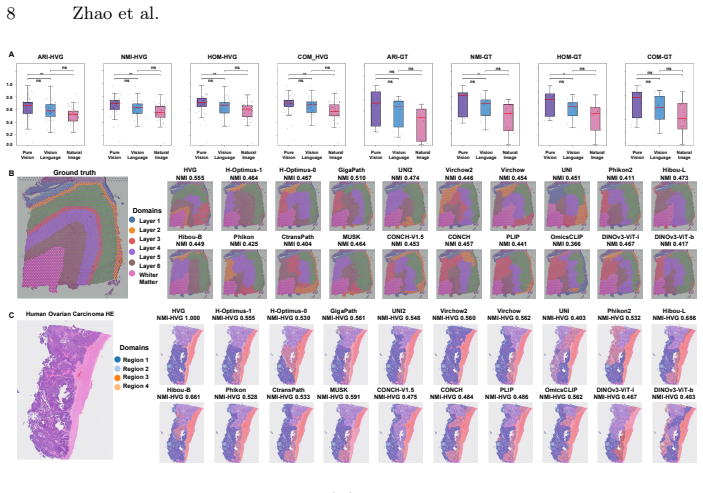
SpaPath-Bench formulates spatial domain identification on paired whole slide image and spatial transcriptomics data as a diagnostic task. It evaluates 19 encoders and seven identification methods across 42 public paired slides, scoring results with unsupervised spatial coherence, transcriptomics-referenced agreement, and expert-referenced agreement. Across 83K runs the benchmark shows that different pretraining paradigms capture distinct aspects of tissue spatial architecture.
What carries the argument
SpaPath-Bench, a representation-level benchmark that turns spatial domain identification on paired whole slide image and spatial transcriptomics data into a diagnostic task for model embeddings.
If this is right
- Different pretraining paradigms produce measurably different abilities to capture tissue spatial architecture.
- The benchmark supplies concrete guidance for choosing or designing pathology foundation models that better respect spatial structure.
- Representation-level tests complement existing clinical-endpoint evaluations by revealing what the embeddings encode about space.
- Models can now be compared directly on their capacity to distinguish meaningful tissue regions and their spatial relationships.
Where Pith is reading between the lines
- Developers could pick pretraining strategies according to the spatial properties needed for a given clinical use case.
- Extending the benchmark to additional paired data types could sharpen distinctions among pretraining approaches.
- Clinical pipelines might add spatial-representation checks alongside accuracy metrics when selecting a model.
Load-bearing premise
That performance on spatial domain identification using paired whole slide images and spatial transcriptomics data measures the spatial representation capability inside the embeddings.
What would settle it
An experiment showing that high scores on SpaPath-Bench metrics do not correspond to better performance on downstream spatial tissue analysis tasks would indicate the benchmark does not capture the intended capability.
Figures

read the original abstract
Pathology foundation models (PFMs) have emerged as a core approach for learning transferable representations from whole slide images (WSIs), and they are typically benchmarked through downstream clinical endpoints. While such task level evaluations are indispensable, they offer limited insight into what the representations themselves encode, particularly whether PFM embeddings can distinguish meaningful tissue regions and capture their spatial relationships. We present SpaPath-Bench, a representation level benchmark designed to diagnose spatial representation capability in PFMs. SpaPath-Bench formulates spatial domain identification (SDI) on paired whole slide image and spatial transcriptomics (ST) data as a diagnostic task. It curates 42 public paired WSI and ST slides, enables large scale evaluation across 19 encoders and seven SDI methods, and measures partition quality using three complementary criteria: unsupervised spatial coherence, transcriptomics referenced agreement, and expert referenced agreement. Across 83K runs, SpaPath-Bench reveals that different pretraining paradigms capture distinct aspects of tissue spatial architecture, and it provides practical guidance for building the next generation of spatially aware computational pathology models. Code and data pipelines are publicly available at https://bokai-zhao.github.io/SpaPath-benchboard/.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SpaPath-Bench, a representation-level benchmark for pathology foundation models (PFMs) that formulates spatial domain identification (SDI) on 42 curated paired WSI-ST slides as a diagnostic task. It evaluates 19 encoders using 7 SDI methods across 83K runs and measures partition quality via three criteria (unsupervised spatial coherence, transcriptomics-referenced agreement, expert-referenced agreement), concluding that different pretraining paradigms capture distinct aspects of tissue spatial architecture and offering guidance for spatially aware models. Code and data pipelines are released publicly.
Significance. If the evaluation isolates PFM spatial inductive bias, the benchmark supplies a useful complement to clinical-endpoint evaluations and the public code release supports reproducibility. The scale (83K runs) and multi-criterion design are strengths that could inform next-generation model development in computational pathology.
major comments (2)
- [Abstract] Abstract: the central claim that the three agreement criteria diagnose 'spatial representation capability encoded in PFM embeddings' rests on the untested assumption that SDI scores are driven by the embeddings' spatial structure rather than ST modality properties, the choice of the seven SDI algorithms, or the 42-slide curation. No ablation replacing PFM embeddings with non-spatial baselines (raw patch statistics or shuffled coordinates) is described, which is load-bearing for the claim that the benchmark measures the intended property.
- [Methods (SDI formulation)] The manuscript does not report whether SDI performance remains high under non-spatial controls, leaving open the possibility that the observed differences across pretraining paradigms reflect interactions with ST data characteristics rather than distinct spatial inductive biases in the embeddings.
minor comments (2)
- [Abstract] The abstract states the scale (83K runs, 19 encoders) but provides no quantitative summary of the main findings (e.g., which paradigms excelled on which criterion); adding one or two key numbers would improve clarity.
- [Results] Notation for the three agreement criteria could be introduced earlier and used consistently when reporting results.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review. The comments correctly identify a missing validation step for isolating spatial inductive bias in the embeddings. We address each major comment below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that the three agreement criteria diagnose 'spatial representation capability encoded in PFM embeddings' rests on the untested assumption that SDI scores are driven by the embeddings' spatial structure rather than ST modality properties, the choice of the seven SDI algorithms, or the 42-slide curation. No ablation replacing PFM embeddings with non-spatial baselines (raw patch statistics or shuffled coordinates) is described, which is load-bearing for the claim that the benchmark measures the intended property.
Authors: We agree that explicit non-spatial controls are necessary to substantiate the central claim. While the current design compares 19 PFMs on identical ST data and SDI methods (thereby attributing relative differences to embedding properties), absolute performance could still be influenced by ST characteristics. In the revision we will add ablations that replace PFM embeddings with non-spatial baselines (random vectors, shuffled coordinates, and raw patch statistics) and report the resulting SDI scores under all three agreement criteria. This will directly test whether high performance requires spatially structured embeddings. revision: yes
-
Referee: [Methods (SDI formulation)] The manuscript does not report whether SDI performance remains high under non-spatial controls, leaving open the possibility that the observed differences across pretraining paradigms reflect interactions with ST data characteristics rather than distinct spatial inductive biases in the embeddings.
Authors: We concur that the absence of these controls leaves the interpretation of paradigm-specific differences open to the concern raised. The revision will therefore include the same non-spatial baseline experiments described above, applied uniformly across the seven SDI methods. Results will be presented in a new methods subsection and supplementary tables so that readers can verify that performance differences across pretraining paradigms are not explained by ST data properties alone. revision: yes
Circularity Check
No circularity: benchmark uses external paired data and standard metrics
full rationale
The paper curates 42 public paired WSI-ST slides and runs 83K evaluations of 19 existing encoders across 7 SDI methods, measuring partition quality via unsupervised coherence, transcriptomics agreement, and expert agreement. No derivation, equation, or claim reduces by construction to author-fitted parameters, self-definitions, or self-citation chains. The central empirical findings about pretraining paradigms rest on independent external data and are not forced by the benchmark design itself.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Science 381(6657), eabq4964 (2023)
Bressan, D., Battistoni, G., Hannon, G.J.: The dawn of spatial omics. Science 381(6657), eabq4964 (2023)
2023
-
[2]
Nature Communications16(1), 3640 (2025)
Campanella, G., Chen, S., Singh, M., Verma, R., Muehlstedt, S., Zeng, J., Stock, A., Croken, M., Veremis, B., Elmas, A., et al.: A clinical benchmark of public self- supervised pathology foundation models. Nature Communications16(1), 3640 (2025)
2025
-
[3]
Nature medicine30(3), 850–862 (2024)
Chen, R.J., Ding, T., Lu, M.Y., Williamson, D.F., Jaume, G., Song, A.H., Chen, B., Zhang, A., Shao, D., Shaban, M., et al.: Towards a general-purpose foundation model for computational pathology. Nature medicine30(3), 850–862 (2024)
2024
-
[4]
Nature Methods22(7), 1568–1582 (2025)
Chen, W., Zhang, P., Tran, T.N., Xiao, Y., Li, S., Shah, V.V., Cheng, H., Bran- nan, K.W., Youker, K., Lai, L., et al.: A visual–omics foundation model to bridge histopathology with spatial transcriptomics. Nature Methods22(7), 1568–1582 (2025)
2025
-
[5]
Nature communications 13(1), 1739 (2022)
Dong, K., Zhang, S.: Deciphering spatial domains from spatially resolved transcrip- tomics with an adaptive graph attention auto-encoder. Nature communications 13(1), 1739 (2022)
2022
- [6]
-
[7]
Nature methods18(11), 1342–1351 (2021)
Hu, J., Li, X., Coleman, K., Schroeder, A., Ma, N., Irwin, D.J., Lee, E.B., Shi- nohara, R.T., Li, M.: Spagcn: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convo- lutional network. Nature methods18(11), 1342–1351 (2021)
2021
-
[8]
Nature medicine29(9), 2307–2316 (2023)
Huang, Z., Bianchi, F., Yuksekgonul, M., Montine, T.J., Zou, J.: A visual–language foundation model for pathology image analysis using medical twitter. Nature medicine29(9), 2307–2316 (2023)
2023
-
[9]
Science384(6698), eadh1938 (2024) 10 Zhao et al
Huuki-Myers, L.A., Spangler, A., Eagles, N.J., Montgomery, K.D., Kwon, S.H., Guo, B., Grant-Peters, M., Divecha, H.R., Tippani, M., Sriworarat, C., et al.: A data-driven single-cell and spatial transcriptomic map of the human prefrontal cortex. Science384(6698), eadh1938 (2024) 10 Zhao et al
2024
-
[10]
Nature Computational Science2(6), 399–408 (2022)
Li, J., Chen, S., Pan, X., Yuan, Y., Shen, H.B.: Cell clustering for spatial tran- scriptomics data with graph neural networks. Nature Computational Science2(6), 399–408 (2022)
2022
-
[11]
Nature Methods pp
Liu,Y.,Wang,C.,Wang,Z.,Chen,L.,Li,Z.,Song,J.,Zou,Q.,Gao,R.,Qian,B.Z., Feng, X., et al.: High-parameter spatial multi-omics through histology-anchored integration. Nature Methods pp. 1–14 (2025)
2025
-
[12]
Nature communications 14(1), 1155 (2023)
Long, Y., Ang, K.S., Li, M., Chong, K.L.K., Sethi, R., Zhong, C., Xu, H., Ong, Z., Sachaphibulkij, K., Chen, A., et al.: Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with graphst. Nature communications 14(1), 1155 (2023)
2023
-
[13]
Nature medicine30(3), 863–874 (2024)
Lu, M.Y., Chen, B., Williamson, D.F., Chen, R.J., Liang, I., Ding, T., Jaume, G., Odintsov, I., Le, L.P., Gerber, G., et al.: A visual-language foundation model for computational pathology. Nature medicine30(3), 863–874 (2024)
2024
-
[14]
Hibou: A Family of Foundational Vision Transformers for Pathology,
Nechaev, D., Pchelnikov, A., Ivanova, E.: Hibou: A family of foundational vision transformers for pathology. arXiv preprint arXiv:2406.05074 (2024)
-
[15]
Nature biomedical engineering pp
Neidlinger, P., El Nahhas, O.S., Muti, H.S., Lenz, T., Hoffmeister, M., Brenner, H., van Treeck, M., Langer, R., Dislich, B., Behrens, H.M., et al.: Benchmark- ing foundation models as feature extractors for weakly supervised computational pathology. Nature biomedical engineering pp. 1–11 (2025)
2025
-
[16]
Nature communications 14(1), 7739 (2023)
Pham, D., Tan, X., Balderson, B., Xu, J., Grice, L.F., Yoon, S., Willis, E.F., Tran, M., Lam, P.Y., Raghubar, A., et al.: Robust mapping of spatiotemporal trajectories and cell–cell interactions in healthy and diseased tissues. Nature communications 14(1), 7739 (2023)
2023
-
[17]
Nature596(7871), 211–220 (2021)
Rao, A., Barkley, D., França, G.S., Yanai, I.: Exploring tissue architecture using spatial transcriptomics. Nature596(7871), 211–220 (2021)
2021
-
[18]
Nature communications13(1), 4076 (2022)
Ren, H., Walker, B.L., Cang, Z., Nie, Q.: Identifying multicellular spatiotemporal organization of cells with spaceflow. Nature communications13(1), 4076 (2022)
2022
-
[19]
Siméoni, O., Vo, H.V., Seitzer, M., Baldassarre, F., Oquab, M., Jose, C., Khalidov, V., Szafraniec, M., Yi, S., Ramamonjisoa, M., Massa, F., Haziza, D., Wehrstedt, L., Wang, J., Darcet, T., Moutakanni, T., Sentana, L., Roberts, C., Vedaldi, A., Tolan, J., Brandt, J., Couprie, C., Mairal, J., Jégou, H., Labatut, P., Bojanowski, P.: DINOv3 (2025),https://ar...
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[20]
Nature Reviews Bioengineering1(12), 930–949 (2023)
Song, A.H., Jaume, G., Williamson, D.F., Lu, M.Y., Vaidya, A., Miller, T.R., Mah- mood, F.: Artificial intelligence for digital and computational pathology. Nature Reviews Bioengineering1(12), 930–949 (2023)
2023
-
[21]
Scientific reports9(1), 5233 (2019)
Traag, V.A., Waltman, L., Van Eck, N.J.: From louvain to leiden: guaranteeing well-connected communities. Scientific reports9(1), 5233 (2019)
2019
-
[22]
Na- ture medicine30(10), 2924–2935 (2024)
Vorontsov, E., Bozkurt, A., Casson, A., Shaikovski, G., Zelechowski, M., Sever- son, K., Zimmermann, E., Hall, J., Tenenholtz, N., Fusi, N., et al.: A foundation model for clinical-grade computational pathology and rare cancers detection. Na- ture medicine30(10), 2924–2935 (2024)
2024
-
[23]
Nature Communications16(1), 1544 (2025)
Wang, C., Chan, A.S., Fu, X., Ghazanfar, S., Kim, J., Patrick, E., Yang, J.Y.: Benchmarkingthetranslationalpotentialofspatialgeneexpressionpredictionfrom histology. Nature Communications16(1), 1544 (2025)
2025
-
[24]
Nature634(8035), 970–978 (2024)
Wang, X., Zhao, J., Marostica, E., Yuan, W., Jin, J., Zhang, J., Li, R., Tang, H., Wang, K., Li, Y., et al.: A pathology foundation model for cancer diagnosis and prognosis prediction. Nature634(8035), 970–978 (2024)
2024
-
[25]
Genome biology19(1), 15 (2018) Title Suppressed Due to Excessive Length 11
Wolf, F.A., Angerer, P., Theis, F.J.: Scanpy: large-scale single-cell gene expression data analysis. Genome biology19(1), 15 (2018) Title Suppressed Due to Excessive Length 11
2018
-
[26]
Nature638(8051), 769–778 (2025)
Xiang, J., Wang, X., Zhang, X., Xi, Y., Eweje, F., Chen, Y., Li, Y., Bergstrom, C., Gopaulchan, M., Kim, T., et al.: A vision–language foundation model for precision oncology. Nature638(8051), 769–778 (2025)
2025
-
[27]
arXiv preprint arXiv:2504.04045 (2025)
Xiong, C., Chen, H., Sung, J.J.: A survey of pathology foundation model: Progress and future directions. arXiv preprint arXiv:2504.04045 (2025)
-
[28]
Genome Medicine16(1), 12 (2024)
Xu, H., Fu, H., Long, Y., Ang, K.S., Sethi, R., Chong, K., Li, M., Uddamvathanak, R., Lee, H.K., Ling, J., et al.: Unsupervised spatially embedded deep representation of spatial transcriptomics. Genome Medicine16(1), 12 (2024)
2024
-
[29]
Nature630(8015), 181–188 (2024)
Xu, H., Usuyama, N., Bagga, J., Zhang, S., Rao, R., Naumann, T., Wong, C., Gero, Z., González, J., Gu, Y., et al.: A whole-slide foundation model for digital pathology from real-world data. Nature630(8015), 181–188 (2024)
2024
-
[30]
Nature Methods21(4), 712–722 (2024)
Yuan, Z., Zhao, F., Lin, S., Zhao, Y., Yao, J., Cui, Y., Zhang, X.Y., Zhao, Y.: Benchmarking spatial clustering methods with spatially resolved transcriptomics data. Nature Methods21(4), 712–722 (2024)
2024
-
[31]
Zhang, A., Jaume, G., Vaidya, A., Ding, T., Mahmood, F.: Accelerating data processing and benchmarking of ai models for pathology. arXiv preprint arXiv:2502.06750 (2025)
-
[32]
BioRxiv pp
Zong, Y., Yu, T., Wang, X., Wang, Y., Hu, Z., Li, Y.: const: an interpretable multi-modal contrastive learning framework for spatial transcriptomics. BioRxiv pp. 2022–01 (2022)
2022
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.