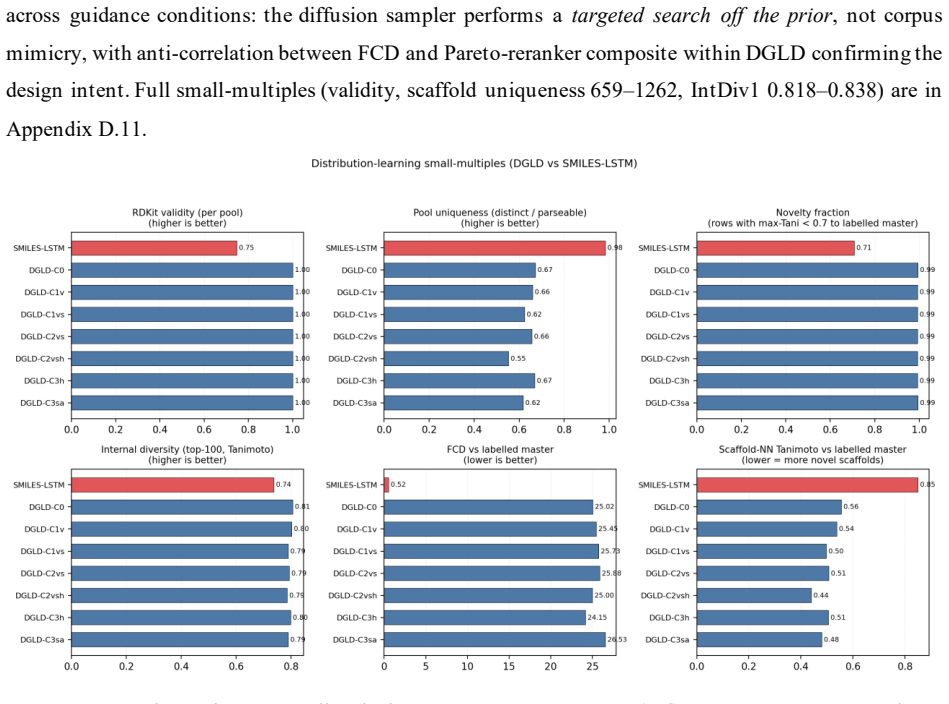
DGLD: Domain-Gated Latent Diffusion for the Discovery of Novel Energetic Materials
Pith reviewed 2026-07-01 16:49 UTC · model grok-4.3
The pith
Domain-gated latent diffusion discovers twelve novel energetic materials validated by first-principles calculations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
DGLD is the only method tested that produces candidates simultaneously novel and on-target when audited with density functional theory, resulting in twelve DFT-confirmed novel energetic material leads from the CHNO space.
What carries the argument
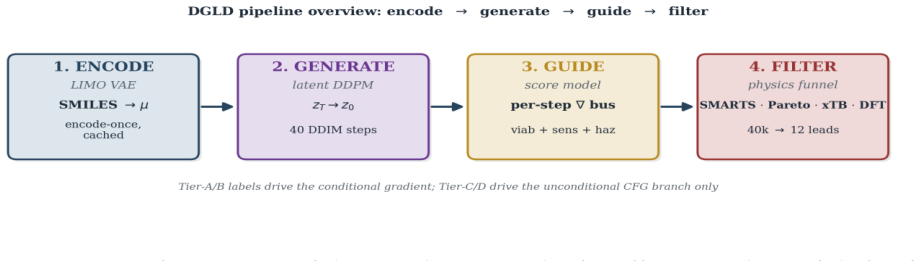
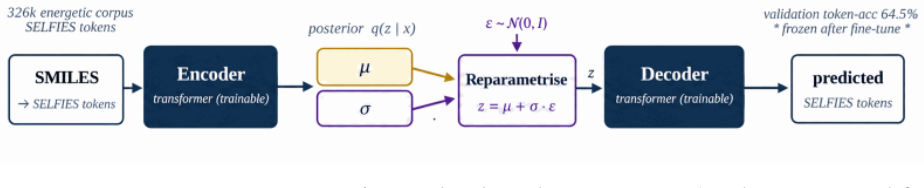
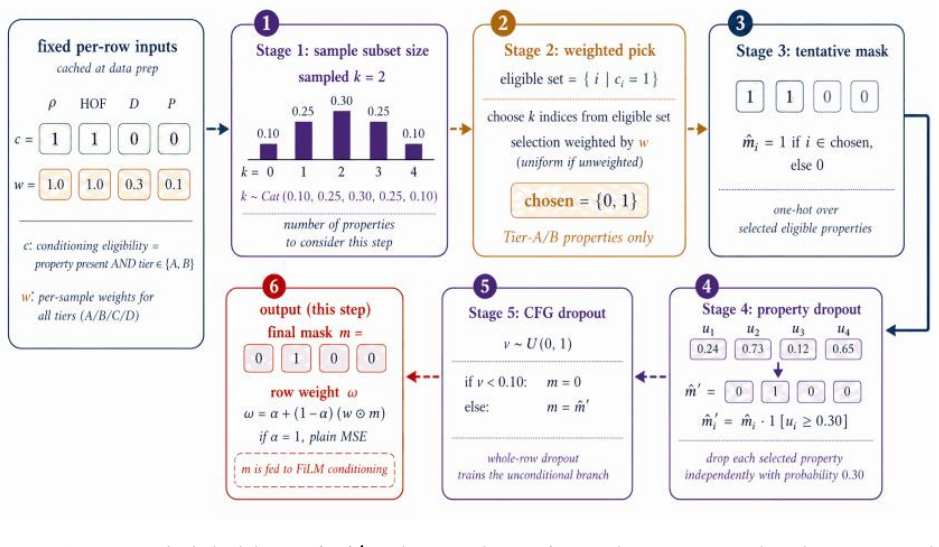
Domain-Gated Latent Diffusion model with label-quality gate at training time, multi-task score-model guidance at sample time, and four-stage chemistry-validation funnel ending in DFT audit.
If this is right
- The next HMX-class energetic material can be discovered, validated, and recommended for synthesis at the cost of a few GPU-days.
- Baseline generative methods either memorize training data at high rates or produce candidates whose performance drops under DFT audit.
- The method can identify leads from disjoint chemotype families with competitive or superior performance metrics.
- High-performance energetic materials become discoverable without relying on manual expert design in the sparse data regime.
Where Pith is reading between the lines
- Gating techniques on label quality could extend to other generative modeling tasks in chemistry where data reliability varies widely.
- Experimental testing of the proposed leads would be needed to confirm that DFT values translate to real material performance.
- The release of mined hard negatives and code may facilitate further improvements or applications in related molecular design problems.
Load-bearing premise
The four-stage chemistry-validation funnel ending in DFT audit correctly identifies materials whose real-world performance will match the calculated values.
What would settle it
Synthesizing the headline compound L1 and experimentally measuring its density and detonation velocity to verify agreement with the DFT predictions of 2.09 g/cm3 and 8.25 km/s.
Figures
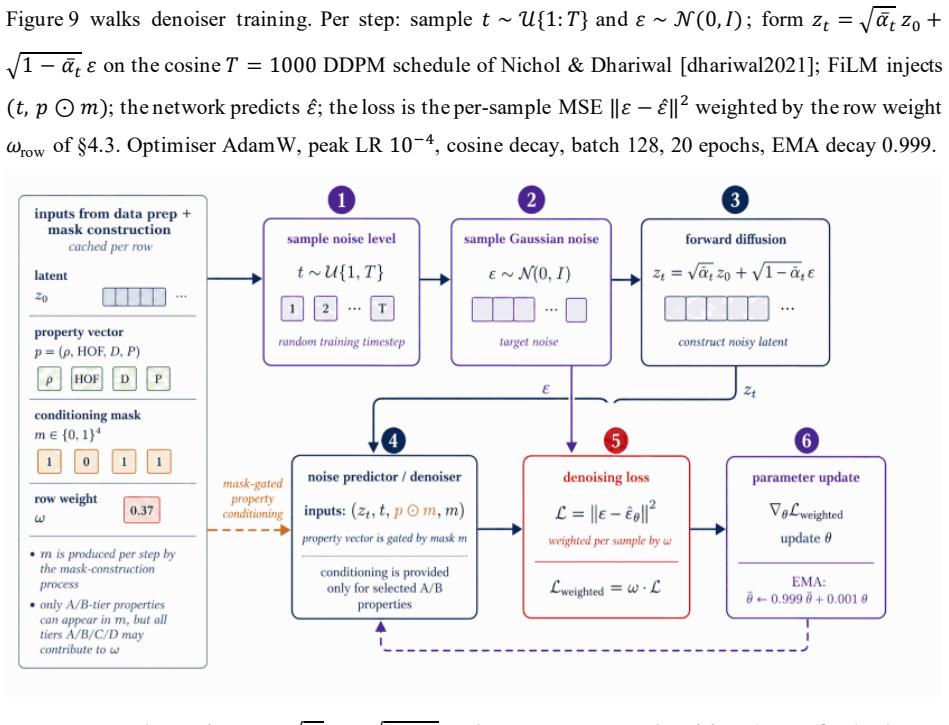

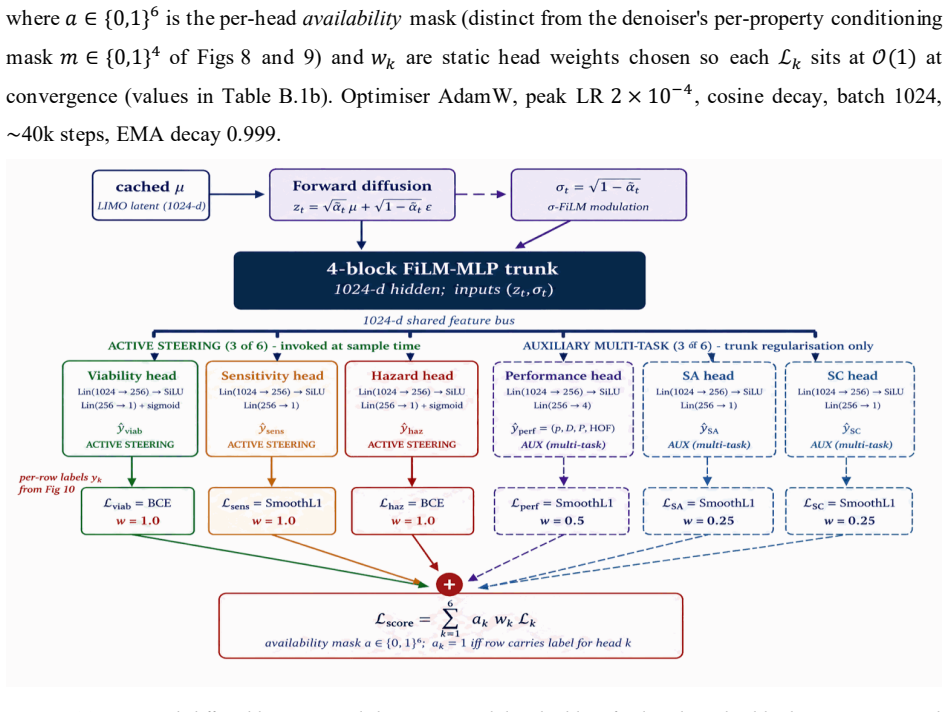


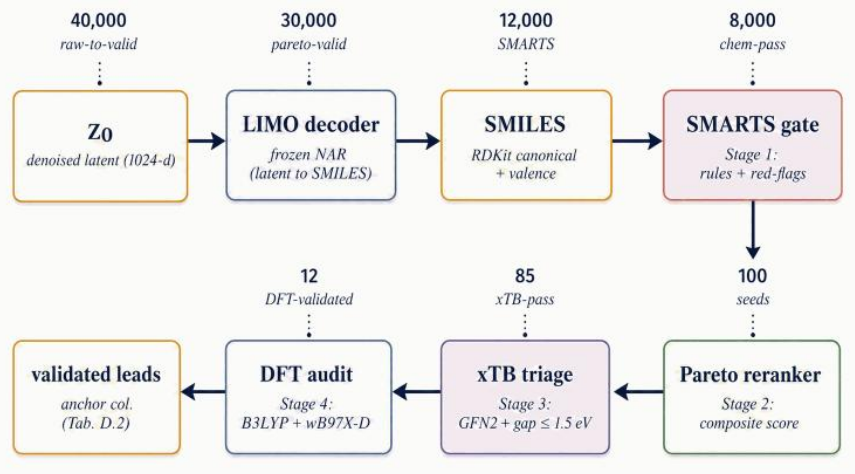
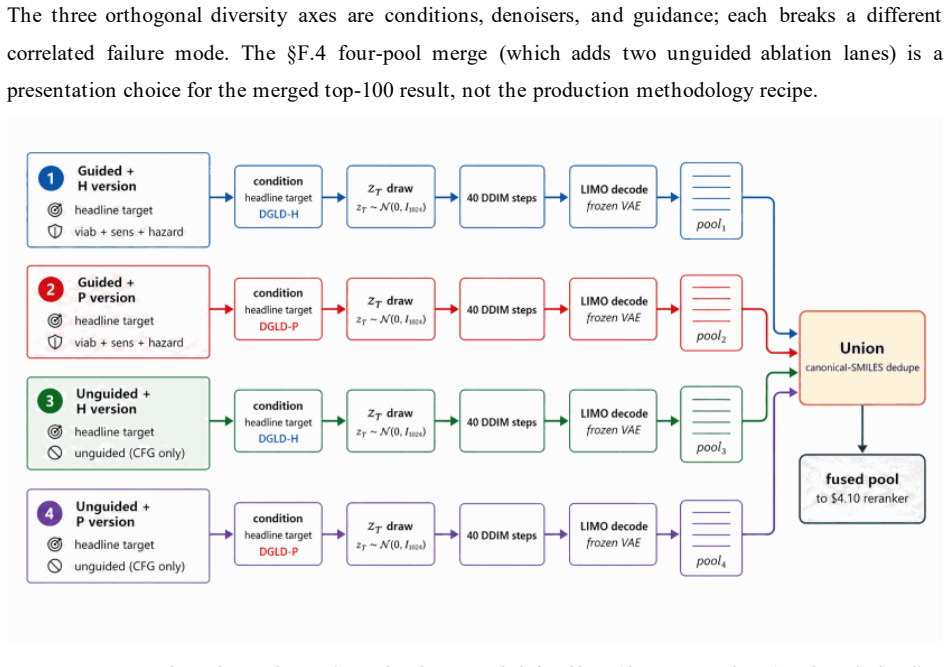

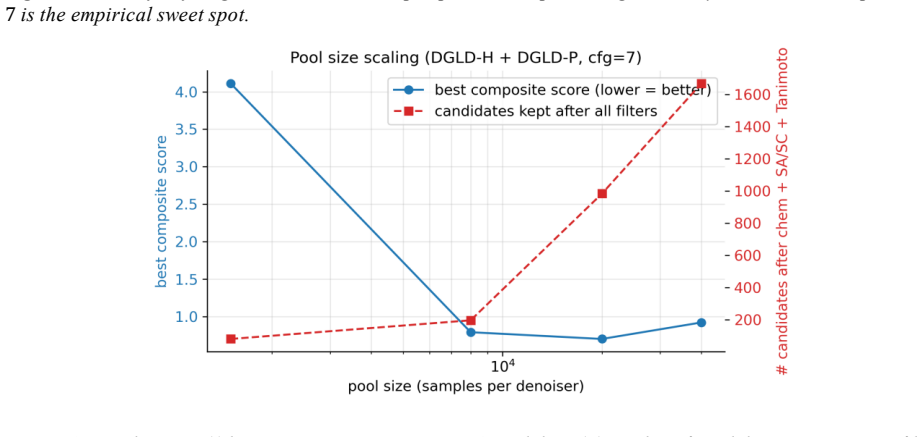

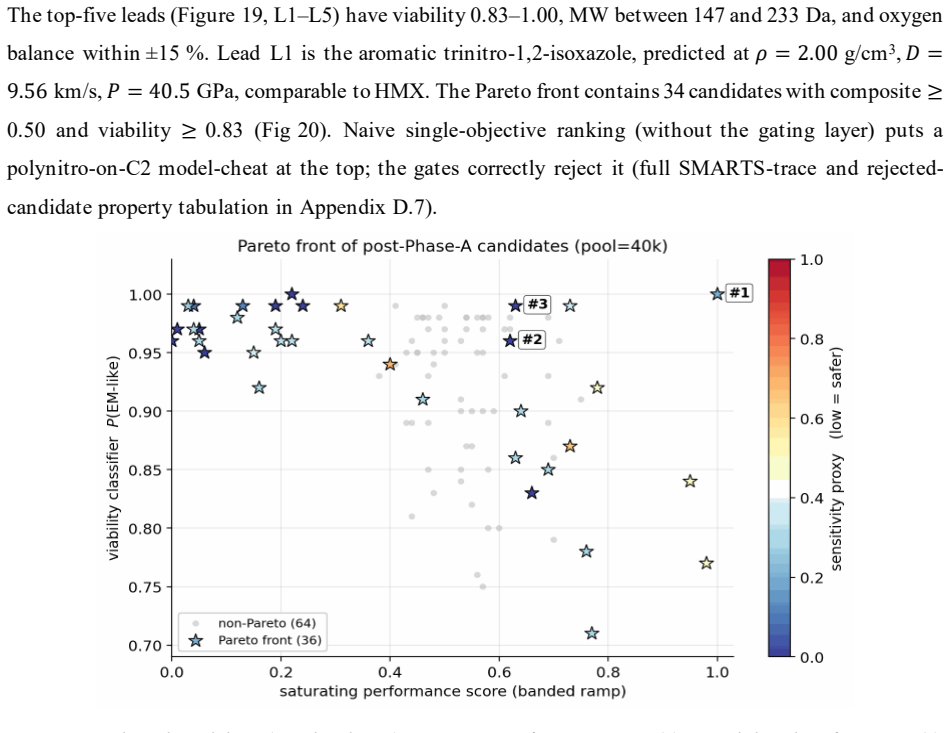
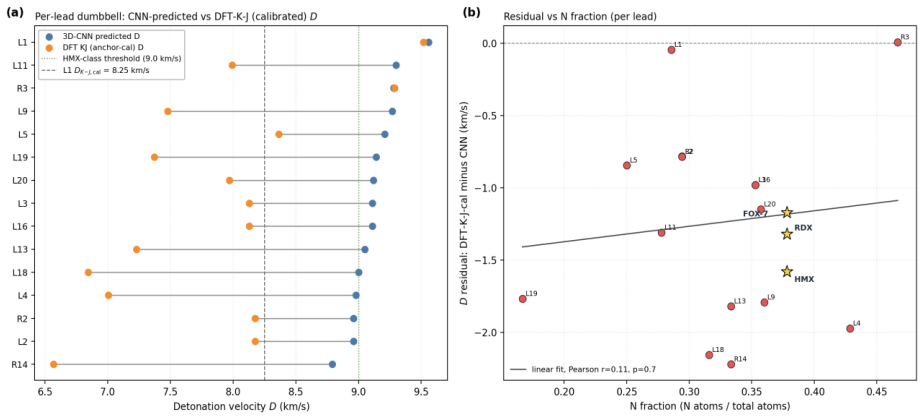
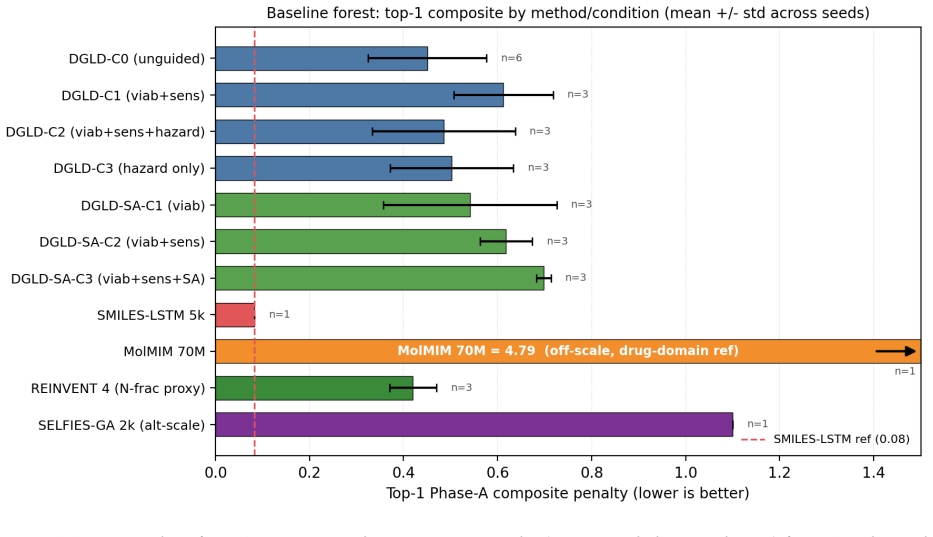


read the original abstract
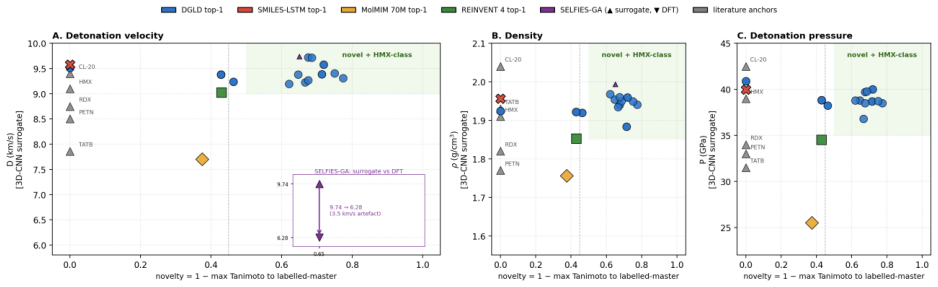
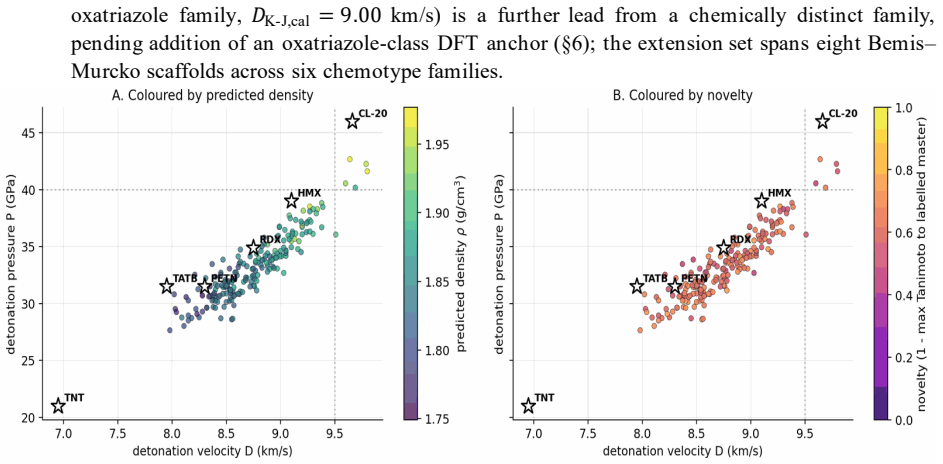
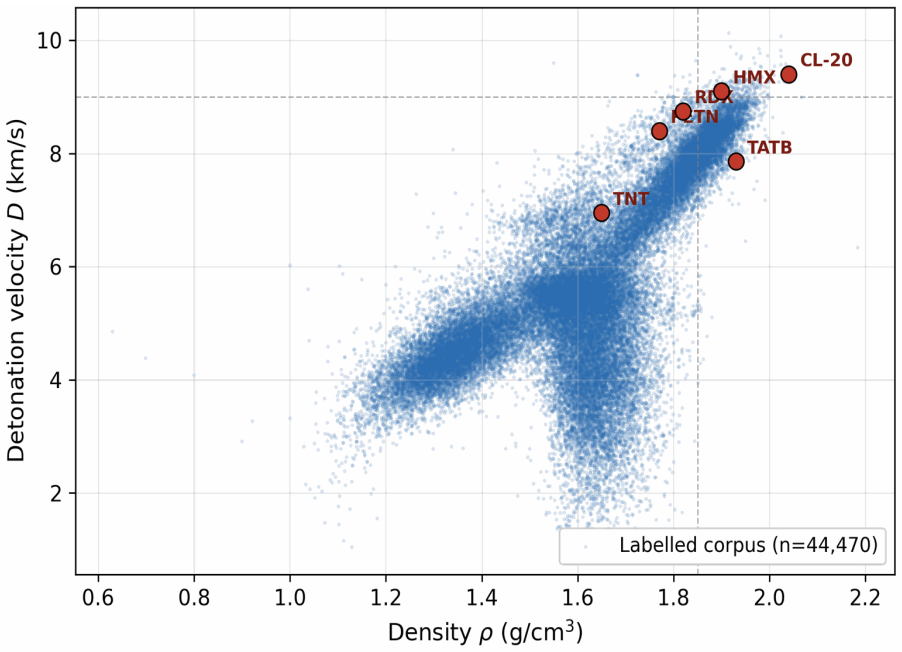
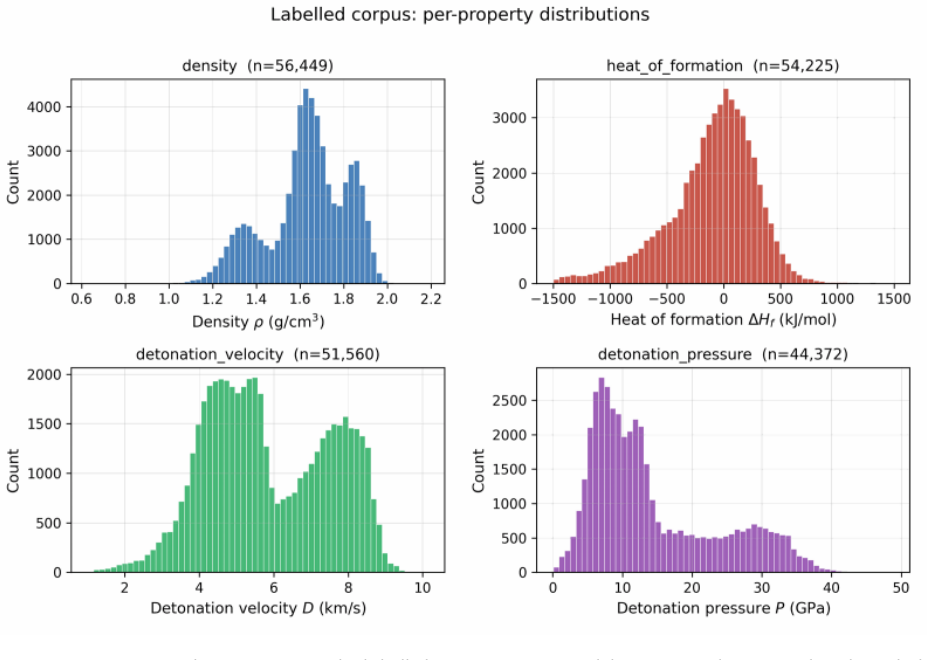
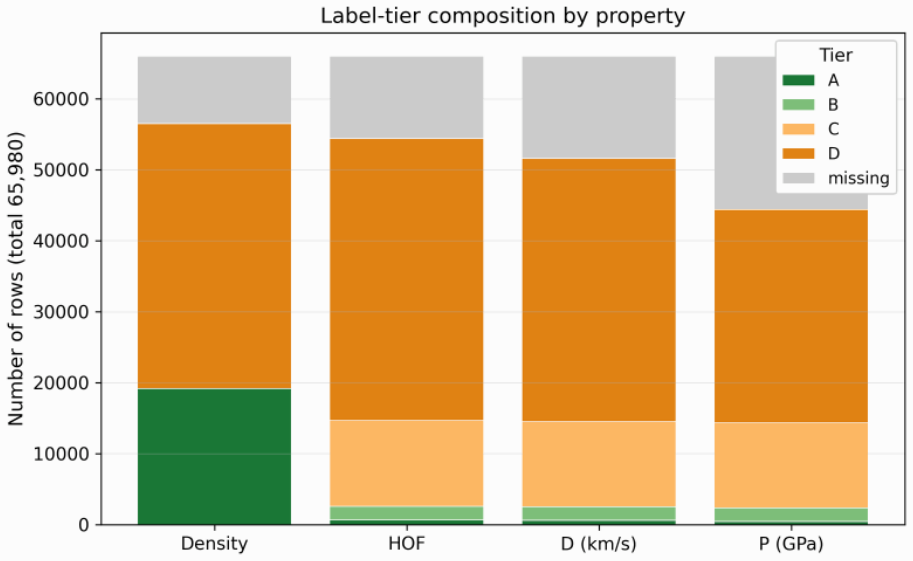
Energetic-materials performance gains translate directly into reduced propellant mass, smaller warheads, and more efficient civilian gas-generators, yet no new HMX-class compound has been disclosed in fifteen years. Designing one is a sparse-label problem: of ~66 k labelled CHNO molecules only ~3 k carry experimental or DFT-quality measurements, and naive generative models trained on the full mixture either memorise the high-performance tail or extrapolate without calibration. We introduce Domain-Gated Latent Diffusion (DGLD): a label-quality gate at training time, multi-task score-model guidance at sample time, and a four-stage chemistry-validation funnel ending in first-principles DFT audit. The result is 12 DFT-confirmed novel leads. The headline compound, 3,4,5-trinitro-1,2-isoxazole (L1), reaches \r{ho}_"cal" =2.09 g/cm3 and D_"K-J,cal" =8.25 km/s and is structurally dissimilar from all 65 980 training molecules (nearest-neighbour Tanimoto 0.27). A co-headline lead, E1 (4-nitro-1,2,3,5-oxatriazole), exceeds L1 on calibrated detonation velocity (D_"K-J,cal" =9.00 km/s) from a chemotype family disjoint from L1's. DGLD is the only method to land in the productive quadrant (simultaneously novel and on-target) at DFT level. SMILES-LSTM memorises 18.3% of its outputs exactly; SELFIES-GA's best novel candidate loses 3.5 km/s under DFT audit; REINVENT 4 generates novel high-N heterocycles but peaks at D=9.02 km/s. Code, checkpoints, and 918 mined hard negatives are released on Zenodo (DOI 10.5281/zenodo.19821953); the next compound to enter the HMX-class band can be discovered, validated, and recommended for synthesis at the cost of a few GPU-days.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces Domain-Gated Latent Diffusion (DGLD) for discovering novel energetic materials in the sparse-label CHNO space (~66k molecules, ~3k with experimental/DFT labels). It employs a label-quality gate during training, multi-task score-model guidance at sampling, and a four-stage chemistry-validation funnel ending in first-principles DFT audit. This produces 12 DFT-confirmed novel leads; the headline compound L1 (3,4,5-trinitro-1,2-isoxazole) reaches ρ_cal=2.09 g/cm³ and D_K-J,cal=8.25 km/s with nearest-neighbour Tanimoto similarity 0.27 to the training set. A second lead E1 reaches D_K-J,cal=9.00 km/s from a disjoint chemotype. DGLD is the only baseline to occupy the productive quadrant (novel and on-target) at DFT level, while SMILES-LSTM memorizes 18.3% of outputs, SELFIES-GA loses 3.5 km/s under DFT, and REINVENT 4 peaks at 9.02 km/s but generates high-N heterocycles. Code, checkpoints, and 918 hard negatives are released on Zenodo.
Significance. If the central claim holds, the work would be significant for providing a practical, calibrated generative framework that navigates the sparse-label regime in energetic-materials design and delivers multiple DFT-audited candidates with performance metrics competitive with or exceeding known high explosives. The explicit release of code, checkpoints, and the mined hard-negative set is a clear strength that supports independent verification of the generative outputs and the funnel.
major comments (1)
- [Abstract / Methods (four-stage funnel)] Abstract and Methods (four-stage funnel description): The headline claim of twelve DFT-confirmed novel leads and DGLD as the sole method in the productive quadrant rests on the four-stage chemistry-validation funnel plus final DFT audit correctly extracting molecules whose computed properties are insensitive to generative artifacts. The manuscript supplies no quantitative stress-test of the funnel against documented diffusion-model failure modes (mode collapse onto high-N heterocycles, density inflation from idealized single-molecule geometries) and no cross-validation of the DFT protocol (functional, basis, dispersion, convergence criteria) on either the 918 hard negatives or the 3 k experimental/DFT reference set.
minor comments (2)
- [Abstract] Abstract: LaTeX formatting artifacts (\r{ho}_"cal", D_"K-J,cal") should be rendered consistently in the published version.
- [Abstract] Abstract: The Tanimoto similarity of 0.27 for L1 is cited as evidence of structural novelty; a short statement of the similarity distribution across the full training set would strengthen the claim.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of the work's significance and for the detailed major comment. We address it directly below.
read point-by-point responses
-
Referee: [Abstract / Methods (four-stage funnel)] Abstract and Methods (four-stage funnel description): The headline claim of twelve DFT-confirmed novel leads and DGLD as the sole method in the productive quadrant rests on the four-stage chemistry-validation funnel plus final DFT audit correctly extracting molecules whose computed properties are insensitive to generative artifacts. The manuscript supplies no quantitative stress-test of the funnel against documented diffusion-model failure modes (mode collapse onto high-N heterocycles, density inflation from idealized single-molecule geometries) and no cross-validation of the DFT protocol (functional, basis, dispersion, convergence criteria) on either the 918 hard negatives or the 3 k experimental/DFT reference set.
Authors: We acknowledge that an explicit quantitative stress-test of the funnel against the cited failure modes would strengthen the presentation. The four-stage funnel was constructed precisely to counter those modes (chemical-validity filter, Tanimoto novelty gate, multi-task score guidance, and final DFT audit), and the empirical outcomes—DGLD alone occupying the productive quadrant while baselines exhibit memorization, property collapse, or high-N heterocycle bias—provide indirect evidence of its effectiveness. The public release of the 918 hard negatives was intended to enable exactly such community-driven stress tests. Regarding the DFT protocol, it follows the same PBE0/def2-TZVP+D3 level used to generate the 3 k reference labels; a dedicated cross-validation subsection on a 200-molecule subset of the reference set and on the hard-negative pool will be added in revision to quantify sensitivity to functional/basis choices. revision: partial
Circularity Check
No significant circularity: claims rest on external DFT validation and independent benchmarks
full rationale
The derivation chain consists of training a domain-gated latent diffusion model on ~66k CHNO molecules (with a label-quality gate), sampling candidates via multi-task guidance, then routing outputs through a four-stage funnel that terminates in first-principles DFT property calculations. Novelty is quantified by Tanimoto distance to the training set (0.27 for L1), and performance is audited by external DFT rather than any internal fitted quantity. Comparisons to SMILES-LSTM, SELFIES-GA and REINVENT 4 are performed on the same DFT protocol and report concrete failure modes (exact memorization, velocity loss, etc.). No step equates a claimed prediction to a fitted parameter by construction, invokes a self-citation as a uniqueness theorem, or renames an input as an output. The central result (12 DFT-confirmed leads, only method in the productive quadrant) is therefore falsifiable by independent DFT runs on the released code and hard-negative set.
Axiom & Free-Parameter Ledger
free parameters (1)
- label-quality gate threshold
axioms (1)
- domain assumption DFT calculations supply sufficiently accurate predictions of density and detonation velocity for CHNO molecules to serve as the final validation standard.
Reference graph
Works this paper leans on
-
[1]
Eckmann, P., Sun, K., Zhao, B., Feng, M., Gilson, M. K., & Yu, R. (2022). LIMO: Latent Inceptionism for Targeted Molecule Generation. ICML 2022. arXiv:2206.09010
-
[2]
Gómez-Bombarelli, R. et al. (2018). Automatic Chemical Design Using a Data -Driven Continuous Representation of Molecules. ACS Central Science 4(2) :268–276. doi:10.1021/acscentsci.7b00572
-
[3]
Jin, W., Barzilay, R., & Jaakkola, T. (2018). Junction Tree Variational Autoencoder for Molecular Graph Generation. ICML 2018. arXiv:1802.04364
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[4]
Reidenbach, D., Livne, M., Ilango, R. K., Gill, M., & Israeli, J. (2023). MolMIM: A Molecular Language Model for Property -Guided Molecule Generation via Mutual Information Machines. (MLDD Workshop, ICLR 2023; arXiv:2208.09016)
-
[5]
Ross, J. et al. (2022). Large -Scale Chemical Language Representations Capture Molecular Structure and Properties. Nature Machine Intelligence 4 :1256 –1264. doi:10.1038/s42256 -022-00580 -7
- [6]
- [7]
- [8]
-
[9]
Irwin, R., Dimitriadis, S., He, J., & Bjerrum, E. J. (2022). Chemformer: A Pre -Trained Transformer for Computational Chemistry. Mach. Learn.: Sci. Tech. 3 :015022. doi:10.1088/2632 -2153/ac3ffb
- [10]
-
[11]
Mathieu, D. (2017). Sensitivity of Energetic Materials: Theoretical Relationships to Detonation Performance and Molecular Structure. Ind. Eng. Chem. Res. 56(31) :8191 –8201. doi:10.1021/acs.iecr.7b02021
-
[12]
Daylight Chemical Information Systems. (2007). SMARTS: A Language for Describing Molecular Patterns. Daylight Theory Manual, Aliso Viejo, CA. daylight.com/dayhtml/doc/theory/theory.smarts.html. SMARTS = SMILES Arbitrary Target Specification: a pattern language extending SMILES that matches molecular substructures, used by RDKit and other cheminformatics t...
2007
-
[13]
Politzer, P., & Murray, J. S. (2014). Some Perspectives on Estimating Detonation Properties of C, H, N, O Compounds. Cent. Eur. J. Energ. Mater. 11(4) :459–474
2014
-
[14]
Sućeska, M. (2018). EXPLO5 v6.05.04 User's Manual. Brodarski Institute, Zagreb, Croatia. Computer program for calculation of detonation parameters from molecular formula, density, and heat of formation via thermochemical -equilibrium Chapman –Jouguet solver with covolume EOS
2018
-
[15]
E., Howard, W
Fried, L. E., Howard, W. M., Souers, P. C., & Vitello, P. A. (2014). Cheetah 7.0 User's Manual. Lawrence Livermore National Laboratory technical report LLNL -SM-664002. Thermochemical -equilibrium detonation code with JCZ3 / BKWS covolume EOS
2014
-
[16]
Kamlet, M. J., & Jacobs, S. J. (1968). Chemistry of Detonations. I. A Simple Method for Calculating Detonation Properties of C -H-N-O Explosives. J. Chem. Phys. 48:23–55. doi:10.1063/1.1667908
-
[17]
C., Boukouvalas, Z., Butrico, M
Elton, D. C., Boukouvalas, Z., Butrico, M. S., Fuge, M. D., & Chung, P. W. (2018). Applying Machine Learning Techniques to Predict the Properties of Energetic Materials. Sci. Rep. 8:9059
2018
-
[18]
D., Son, S
Casey, A. D., Son, S. P., Bilionis, I., & Barnes, B. C. (2020). Prediction of Energetic Material Properties from Electronic Structure Using 3D Convolutional Neural Networks. J. Chem. Inf. Model. 60(10) :4457–
2020
-
[19]
doi:10.1021/acs.jcim.0c00259
-
[20]
Zhou, G. et al. (2023). Uni -Mol: A Universal 3D Molecular Representation Learning Framework. ICLR 2023
2023
-
[21]
Huang, X. et al. (2021). Applying Machine Learning to Balance Performance and Stability of High Energy Density Materials. iScience 24 :102803
2021
-
[22]
Hervé, G., Roussel, C., & Graindorge, H. (2010). Selective Preparation of 3,4,5 -Trinitro-1H-pyrazole: A Stable All-Carbon-Substituted Trinitro Heterocycle, and Related Trinitroisoxazole Chemistry. Angew. Chem. Int. Ed. 49(18) :3177 –3181. doi:10.1002/anie.201000764. 47
-
[23]
Sabatini, J. J. (2018). A Review of Nitroisoxazole -Based Energetic Compounds. Propellants, Explosives, Pyrotechnics 43(1) :28–37. doi:10.1002/prep.201700225
-
[24]
Konnov, A. A., Lisyutkin, A. D., Vinogradov, D. B., Nazarova, A. A., Pivkina, A. N., & Fershtat, L. L. (2025). Synthesis of 4 -Nitroisoxazole-Based Energetic Materials. Org. Lett. 27(14) :3795–3799. doi:10.1021/acs.orglett.5c01074
-
[25]
Ho, J., & Salimans, T. (2022). Classifier -Free Diffusion Guidance. arXiv:2207.12598
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[26]
Dhariwal, P., & Nichol, A. (2021). Diffusion Models Beat GANs on Image Synthesis. NeurIPS 2021. arXiv:2105.05233
work page internal anchor Pith review Pith/arXiv arXiv 2021
-
[27]
Song, Y., & Ermon, S. (2019). Generative Modeling by Estimating Gradients of the Data Distribution. NeurIPS 2019 . arXiv:1907.05600
work page internal anchor Pith review Pith/arXiv arXiv 2019
-
[28]
Song, Y. et al. (2021). Score -Based Generative Modeling through Stochastic Differential Equations. ICLR
2021
-
[29]
Ho, J., Jain, A., & Abbeel, P. (2020). Denoising Diffusion Probabilistic Models. NeurIPS 2020 . arXiv:2006.11239
work page internal anchor Pith review Pith/arXiv arXiv 2020
-
[30]
Rombach, R., Blattmann, A., Lorenz, D., Esser, P., & Ommer, B. (2022). High-Resolution Image Synthesis with Latent Diffusion Models. CVPR 2022. arXiv:2112.10752
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[31]
Krenn, M., Häse, F., Nigam, A., Friederich, P., & Aspuru -Guzik, A. (2020). Self-Referencing Embedded Strings (SELFIES): A 100% Robust Molecular String Representation. Mach. Learn.: Sci. Tech. 1 :045024
2020
-
[32]
Ertl, P., & Schuffenhauer, A. (2009). Estimation of Synthetic Accessibility Score of Drug-Like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 1:8
2009
-
[33]
W., Rogers, L., Green, W
Coley, C. W., Rogers, L., Green, W. H., & Jensen, K. F. (2018). SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 58(2) :252–261
2018
-
[34]
J., & Tanimoto, T
Rogers, D. J., & Tanimoto, T. T. (1960). A Computer Program for Classifying Plants. Science 132(3434) :1115 –1118
1960
-
[35]
RDKit: Open -source cheminformatics
Landrum, G., & contributors. RDKit: Open -source cheminformatics. rdkit.org
-
[36]
Sterling, T., & Irwin, J. J. (2015). ZINC 15: Ligand Discovery for Everyone. J. Chem. Inf. Model. 55(11) :2324 –2337
2015
-
[37]
Kim, S. et al. (2023). PubChem 2023 Update. Nucleic Acids Res. 51(D1) :D1373–D1380
2023
- [38]
-
[40]
H., He, J., Tibo, A., Janet, J
Loeffler, H. H., He, J., Tibo, A., Janet, J. P., Voronov, A., Mervin, L. H., & Engkvist, O. (2024). REINVENT 4: Modern AI -driven generative molecule design. J. Cheminformatics 16 :20. doi:10.1186/s13321-024- 00812 -5
-
[41]
Yang, X., Zhang, J., Yoshizoe, K., Terayama, K., & Tsuda, K. (2017). ChemTS: An efficient python library for de novo molecular generation. Sci. Tech. Adv. Mater. 18(1) :972–976. doi:10.1080/14686996.2017.1401424
-
[42]
Bagal, V., Aggarwal, R., Vinod, P. K., & Priyakumar, U. D. (2022). MolGPT: Molecular Generation Using a Transformer -Decoder Model. J. Chem. Inf. Model. 62(9) :2064 –2076. doi:10.1021/acs.jcim.1c00600
-
[43]
Winter, R., Montanari, F., Noé, F., & Clevert, D.-A. (2019). Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 10(6) :1692–1701. doi:10.1039/C8SC04175J
- [45]
- [46]
- [47]
- [48]
-
[49]
Nefati, H., Cense, J.-M., & Legendre, J.-J. (1996). Prediction of the Impact Sensitivity by Neural Networks. J. Chem. Inf. Comput. Sci. 36(4) :804–810. doi:10.1021/ci950223m
-
[50]
Klapötke, T. M. Chemistry of High -Energy Materials , 5th ed. (de Gruyter, 2019). doi:10.1515/9783110624571
-
[51]
Griffiths, R. -R., & Hernández -Lobato, J. M. (2020). Constrained Bayesian optimization for automatic chemical design using variational autoencoders. Chem. Sci. 11(2) :577–586. doi:10.1039/C9SC04026A
-
[52]
Yang, K. et al. (2019). Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 59(8) :3370 –3388. doi:10.1021/acs.jcim.9b00237
-
[53]
Schütt, K. T., Sauceda, H. E., Kindermans, P.-J., Tkatchenko, A., & Müller, K.-R. (2018). SchNet: A deep learning architecture for molecules and materials. J. Chem. Phys. 148:241722. doi:10.1063/1.5019779
-
[54]
Qiao, Z., Welborn, M., Anandkumar, A., Manby, F. R., & Miller III, T. F. (2020). OrbNet: Deep learning for quantum chemistry using symmetry -adapted atomic -orbital features. J. Chem. Phys. 153 :124111. doi:10.1063/5.0021955
-
[55]
Brown, N., Fiscato, M., Segler, M. H. S., & Vaucher, A. C. (2019). GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 59(3) :1096 –1108. doi:10.1021/acs.jcim.8b00839
-
[56]
Polykovskiy, D. et al. (2020). Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Frontiers in Pharmacology 11 :565644. doi:10.3389/fphar.2020.565644
-
[57]
Preuer, K., Renz, P., Unterthiner, T., Hochreiter, S., & Klambauer, G. (2018). Fréchet ChemNet Distance: A Metric for Generative Models for Molecules in Drug Discovery. J. Chem. Inf. Model. 58(9) :1736 –1741
2018
-
[58]
Reymond, J.-L. (2015). The chemical space project. Acc. Chem. Res. 48(3) :722–730
2015
-
[59]
Hand-compilation of measured density, heat of formation, and detonation properties for ~3 000 known energetic CHNO compounds, assembled in this work from secondary literature compilations: Klapötke, T. M. Chemistry of High -Energy Materials , 5th ed. (de Gruyter, 2019); Cooper, P. W. Explosives Engineering (Wiley-VCH, 1996); and Dobratz, B. M. & Crawford,...
2019
-
[60]
cameochemicals.noaa.gov
NIST CAMEO Chemicals: Database of Hazardous Materials and Reactivity. cameochemicals.noaa.gov
-
[61]
dangerous reactivity
Bruns, H., & Watson, P. (2020). SMARTS-based reactivity demerit catalogues for energetic-materials triage (in-house compilation following the ChemAxon “dangerous reactivity” rule set)
2020
-
[62]
Bannwarth, C., Ehlert, S., & Grimme, S. (2019). GFN2-xTB: An Accurate and Broadly Parametrized Self- Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 15(3) :1652 –1671. doi:10.1021/acs.jctc.8b01176
-
[63]
Goerigk, L., Hansen, A., Bauer, C., Ehrlich, S., Najibi, A., & Grimme, S. (2017). A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 19(48) :32184–32215. doi:10.1039/C7CP04913G
-
[64]
Bondi, A. (1964). van der Waals Volumes and Radii. J. Phys. Chem. 68(3) :441–451. doi:10.1021/j100785a001
-
[65]
-L., Engkvist, O., & Bjerrum, E
Genheden, S., Thakkar, A., Chadimová, V., Reymond, J. -L., Engkvist, O., & Bjerrum, E. J. (2020). AiZynthFinder: a fast, robust and flexible open -source software for retrosynthetic planning. Journal of Cheminformatics 12 :70. doi:10.1186/s13321 -020-00472 -1
-
[66]
Sun, Q., Zhang, X., Banerjee, S., Bao, P., et al. (2020). Recent developments in the PySCF program package. J. Chem. Phys. 153:024109. doi:10.1063/5.0006074
-
[67]
Perez, E., Strub, F., de Vries, H., Dumoulin, V., & Courville, A. (2018). FiLM: Visual Reasoning with a General Conditioning Layer. AAAI 2018. arXiv:1709.07871
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[68]
Xu, M., Powers, A., Dror, R. O., Ermon, S., & Leskovec, J. (2023). Geometric Latent Diffusion Models for 3D Molecule Generation. ICML 2023. arXiv:2305.01140
-
[69]
Z. et al. (2025). De novo multi -objective generation framework for energetic materials with trading off energy and stability. npj Computational Materials . doi:10.1038/s41524 -025-01845 -6. 49
-
[70]
Choi, J. B., Nguyen, P. C. H., Sen, O., Udaykumar, H. S., & Baek, S. (2023). Artificial Intelligence Approaches for Energetic Materials by Design: State of the Art, Challenges, and Future Directions. Propellants, Explosives, Pyrotechnics 48(4) , e202200276. doi:10.1002/prep.202200276
-
[71]
Arnold, J. E., & Day, G. M. (2023). Crystal Structure Prediction of Energetic Materials. Crystal Growth & Design. doi:10.1021/acs.cgd.3c00706
-
[72]
Davis, J. V., Marrs III, F. W., Cawkwell, M. J., & Manner, V. W. (2024). Machine Learning Models for High Explosive Crystal Density and Performance. Chemistry of Materials 36(22) , 11109 –11118. doi:10.1021/acs.chemmater.4c01978
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.