Computational Modeling of Antibody-Antigen Complexes: PLM-Based and MSA-Based Approaches
Pith reviewed 2026-06-29 09:39 UTC · model grok-4.3
The pith
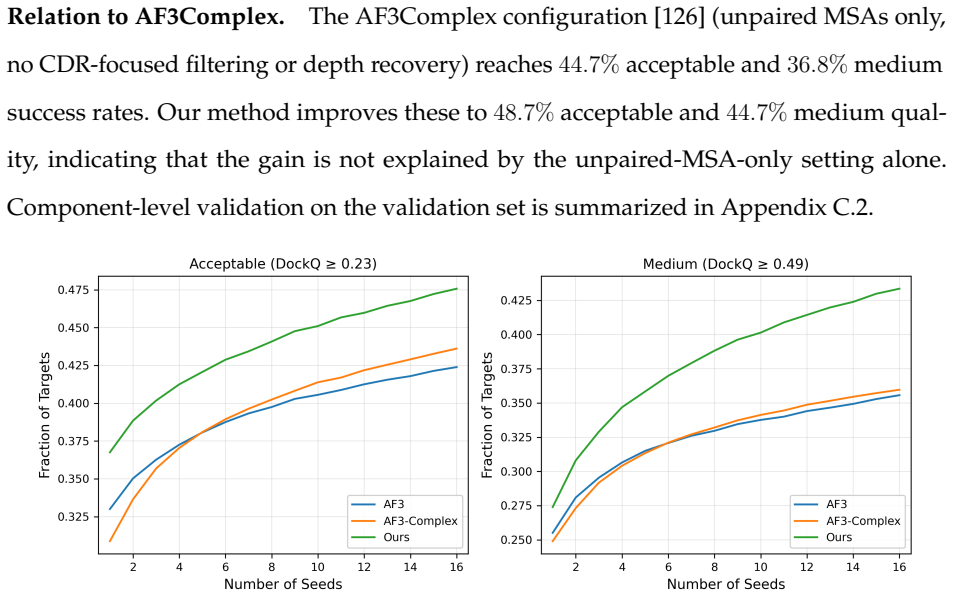
MSA refinement and convergence-aware recycling improve antibody-antigen complex prediction over the AlphaFold3 baseline
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
MSA refinement, which combines CDR-focused filtering with depth recovery from a larger sequence database, and convergence-aware recycling, which selects a stable intermediate recycle state for final diffusion sampling, together provide consistent gains over the AlphaFold3 baseline on a held-out antibody-antigen test set. Because the methods modify MSA construction and recycling behavior rather than model parameters, they apply without retraining or weight access.
What carries the argument
MSA refinement (CDR-focused filtering with depth recovery) and convergence-aware recycling, which alter sequence alignment inputs and the recycling step in diffusion-based complex prediction.
If this is right
- Single-sequence PLM representations do not reliably identify binding interfaces in antibody-antigen complexes without co-evolutionary signals between the partners.
- PLM-based methods achieve the best CDR-H3 accuracy among compared approaches on antibody monomer prediction.
- The interventions deliver gains on antibody-antigen complexes by changing only MSA construction and recycling behavior, without any retraining.
- Accurate computational modeling of antibody-antigen interactions can prioritize candidates and reduce the experimental burden in therapeutic antibody discovery.
Where Pith is reading between the lines
- The same MSA curation steps could be tested on other specialized interfaces where standard alignments perform poorly.
- Ablating the CDR-focused filter versus the depth-recovery step separately would clarify which component drives most of the reported gain.
- If the gains hold on larger or more diverse antibody sets, the approach would support broader use in rational antibody design pipelines.
Load-bearing premise
The held-out antibody-antigen test set is representative of real-world cases and the observed improvements are caused by the MSA refinement and recycling changes rather than other unstated factors in data processing or model behavior.
What would settle it
Running the modified pipeline on an independently assembled antibody-antigen complex test set and finding no accuracy improvement or a reversal of the reported gains would falsify the claim of consistent benefits from the two interventions.
Figures



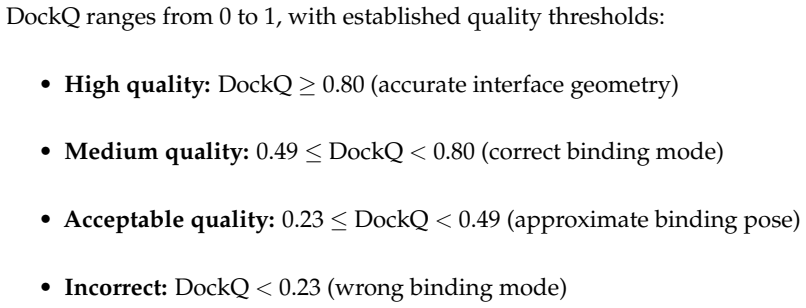
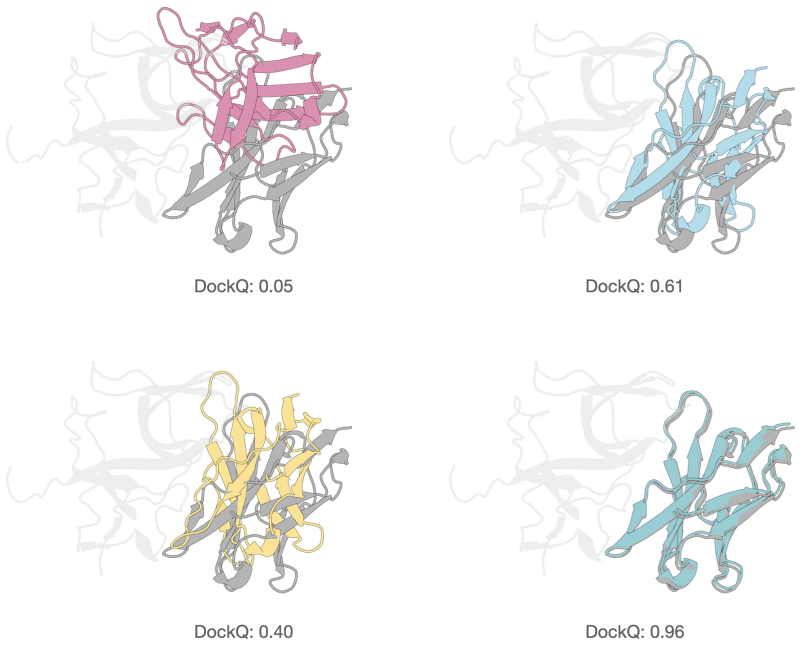









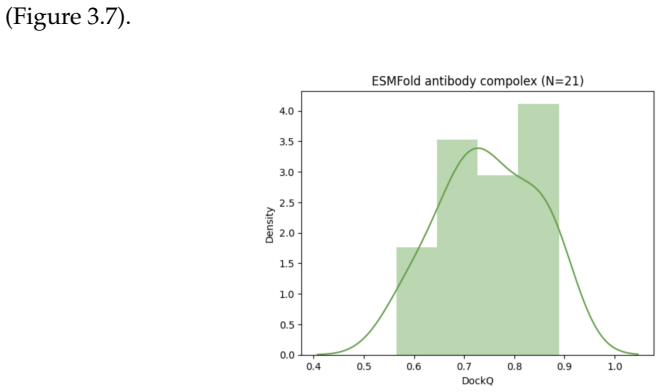











read the original abstract
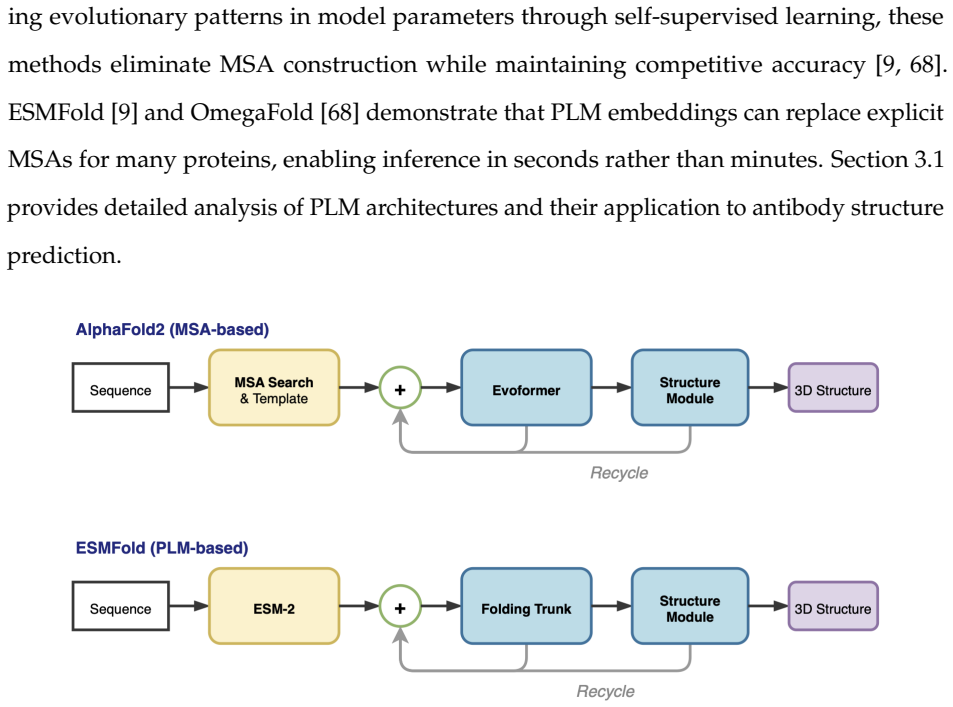
Antibodies play a central role in the immune response by specifically recognizing and neutralizing antigens, and therapeutic antibodies have become major drugs for cancer and autoimmune diseases. However, their discovery still relies on extensive in vitro screening, and accurate computational modeling of antibody structures and antibody-antigen interactions can prioritize candidates, reduce experimental burden, and accelerate rational design. Despite recent advances in high-accuracy protein and complex prediction, a persistent performance gap remains for antibody-related tasks compared with general protein-protein interactions, limiting downstream design. This thesis investigates why antibody-related tasks are harder and proposes improvements along two complementary directions. First, we investigate protein language model (PLM)-based methods for antibody and antibody-antigen structure prediction. Using embeddings from multiple PLMs, our approach achieves the best CDR-H3 accuracy among compared PLM-based methods on antibody monomer prediction. Extending it to complex prediction does not generalize: without co-evolutionary signals between antibody and antigen, single-sequence PLM representations do not reliably identify binding interfaces. Second, we develop two MSA-based interventions for antibody-antigen complex prediction: MSA refinement, which combines CDR-focused filtering with depth recovery from a larger sequence database, and convergence-aware recycling, which selects a stable intermediate recycle state for final diffusion sampling. Together, these interventions provide consistent gains over the AlphaFold3 baseline on a held-out antibody-antigen test set. Because the methods modify MSA construction and recycling behavior rather than model parameters, they apply without retraining or weight access.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript examines difficulties in modeling antibody-antigen complexes and proposes two lines of work. PLM-based methods using embeddings from multiple protein language models achieve the highest CDR-H3 accuracy among compared approaches for antibody monomer prediction, but fail to generalize to complex prediction because they lack co-evolutionary signals between antibody and antigen. For complex prediction the authors introduce two MSA-based interventions—MSA refinement (CDR-focused filtering plus depth recovery from a larger sequence database) and convergence-aware recycling (selection of a stable intermediate recycle state for final diffusion sampling)—that together yield consistent gains over the AlphaFold3 baseline on a held-out antibody-antigen test set. Because the interventions alter only MSA construction and recycling behavior, they require no retraining or weight access.
Significance. If the reported gains are robust, attributable to the two named interventions, and generalize beyond the held-out set, the work would supply immediately usable, training-free improvements to antibody-antigen modeling, an area of clear practical importance for therapeutic design. The parameter-free character of the changes is a genuine strength. However, the absence of any quantitative metrics, ablation results, test-set statistics, or error bars in the manuscript prevents evaluation of whether the claimed improvements are real, reproducible, or merely artifacts of test-set selection or unstated processing choices.
major comments (2)
- [Abstract] Abstract, second paragraph: the central claim that 'MSA refinement … and convergence-aware recycling … together provide consistent gains over the AlphaFold3 baseline' is unsupported by any ablation table, statistical test, or quantitative metric. Without isolating the contribution of each intervention or reporting error bars and significance, attribution of performance differences to the described changes rather than incidental data-processing or sampling differences cannot be verified.
- [Abstract] Abstract, second paragraph: no information is given on the held-out antibody-antigen test set (size, sequence-identity distribution relative to AF3 training data, epitope diversity, or MSA-depth statistics). If the test complexes happen to possess unusually deep MSAs or unusually stable recycle trajectories, the observed improvement could be an artifact of test-set composition rather than a general property of the interventions.
minor comments (1)
- [Abstract] The manuscript would benefit from a short explicit statement of why single-sequence PLM representations cannot capture inter-chain contacts even when monomer accuracy is high.
Simulated Author's Rebuttal
We thank the referee for identifying the need for stronger quantitative support and test-set characterization. We will revise the manuscript to incorporate ablation studies, metrics with error bars and significance tests, and full test-set statistics as detailed below.
read point-by-point responses
-
Referee: [Abstract] Abstract, second paragraph: the central claim that 'MSA refinement … and convergence-aware recycling … together provide consistent gains over the AlphaFold3 baseline' is unsupported by any ablation table, statistical test, or quantitative metric. Without isolating the contribution of each intervention or reporting error bars and significance, attribution of performance differences to the described changes rather than incidental data-processing or sampling differences cannot be verified.
Authors: We agree the abstract claim requires direct supporting evidence. The revised manuscript will add an ablation table isolating MSA refinement and convergence-aware recycling, report mean metrics with standard deviations across replicates, and include statistical tests (e.g., paired t-tests) comparing against the AF3 baseline. These additions will be placed in the results section with a brief reference in the abstract. revision: yes
-
Referee: [Abstract] Abstract, second paragraph: no information is given on the held-out antibody-antigen test set (size, sequence-identity distribution relative to AF3 training data, epitope diversity, or MSA-depth statistics). If the test complexes happen to possess unusually deep MSAs or unusually stable recycle trajectories, the observed improvement could be an artifact of test-set composition rather than a general property of the interventions.
Authors: We agree that test-set details are essential to assess generalizability. The revision will report the test-set size, maximum sequence identity to AF3 training data, epitope diversity summary, and MSA-depth distributions for both antibody and antigen chains. These statistics will be added to the methods or results section to demonstrate the set is representative rather than biased toward deep MSAs or stable trajectories. revision: yes
Circularity Check
No circularity: empirical gains on held-out set are independent of inputs
full rationale
The paper describes two MSA-based interventions (CDR-focused filtering plus depth recovery; convergence-aware recycle selection) and reports consistent gains versus an AlphaFold3 baseline on a held-out antibody-antigen test set. No equations, fitted parameters, or self-citations appear in the provided text that would reduce the measured improvements to the interventions by construction. The methods alter input construction (MSA and recycling) rather than model weights, and evaluation occurs on data external to the training or fitting process, rendering the central claim self-contained.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Structure and function of immunoglobulins
Harry W Schroeder Jr and Lisa Cavacini. Structure and function of immunoglobulins. Journal of allergy and clinical immunology, 125(2):S41–S52, 2010
2010
-
[2]
Five computational developability guidelines for therapeutic antibody profiling.Proceed- ings of the National Academy of Sciences, 116(10):4025–4030, 2019
Matthew IJ Raybould, Claire Marks, Konrad Krawczyk, Bruck Taddese, Jaroslaw Nowak, Alan P Lewis, Alexander Bujotzek, Jiye Shi, and Charlotte M Deane. Five computational developability guidelines for therapeutic antibody profiling.Proceed- ings of the National Academy of Sciences, 116(10):4025–4030, 2019
2019
-
[3]
Improving b-cell epitope prediction and its application to global antibody-antigen docking.Bioinformatics, 30(16):2288–2294, 2014
Konrad Krawczyk, Xiaofeng Liu, Terry Baker, Jiye Shi, and Charlotte M Deane. Improving b-cell epitope prediction and its application to global antibody-antigen docking.Bioinformatics, 30(16):2288–2294, 2014
2014
-
[4]
Computational approaches to therapeutic antibody design: established methods and emerging trends.Briefings in bioinformatics, 21(5):1549–1567, 2020
Richard A Norman, Francesco Ambrosetti, Alexandre MJJ Bonvin, Lucy J Colwell, Sebastian Kelm, Sandeep Kumar, and Konrad Krawczyk. Computational approaches to therapeutic antibody design: established methods and emerging trends.Briefings in bioinformatics, 21(5):1549–1567, 2020
2020
-
[5]
Computa- tional and artificial intelligence-based methods for antibody development.Trends in pharmacological sciences, 44(3):175–189, 2023
Jisun Kim, Matthew McFee, Qiao Fang, Osama Abdin, and Philip M Kim. Computa- tional and artificial intelligence-based methods for antibody development.Trends in pharmacological sciences, 44(3):175–189, 2023
2023
-
[6]
Advances in computational structure-based antibody design.Current opinion in structural biology, 74:102379, 2022
Alissa M Hummer, Brennan Abanades, and Charlotte M Deane. Advances in computational structure-based antibody design.Current opinion in structural biology, 74:102379, 2022
2022
-
[7]
Optimization of therapeutic antibodies by predicting antigen specificity from 111 antibody sequence via deep learning.Nature biomedical engineering, 5(6):600–612, 2021
Derek M Mason, Simon Friedensohn, Cédric R Weber, Christian Jordi, Bastian Wagner, Simon M Meng, Roy A Ehling, Lucia Bonati, Jan Dahinden, Pablo Gainza, et al. Optimization of therapeutic antibodies by predicting antigen specificity from 111 antibody sequence via deep learning.Nature biomedical engineering, 5(6):600–612, 2021
2021
-
[8]
Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
2021
-
[9]
Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379 (6637):1123–1130, 2023
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379 (6637):1123–1130, 2023
2023
-
[10]
Accurate prediction of protein structures and interactions using a three-track neural network.Science, 373(6557):871–876, 2021
Minkyung Baek, Frank DiMaio, Ivan Anishchenko, Justas Dauparas, Sergey Ovchin- nikov, Gyu Rie Lee, Jue Wang, Qian Cong, Lisa N Kinch, R Dustin Schaeffer, et al. Accurate prediction of protein structures and interactions using a three-track neural network.Science, 373(6557):871–876, 2021
2021
-
[11]
Evaluation of alphafold antibody–antigen modeling with implications for improving predictive accuracy.Protein Science, 33(1):e4865, 2024
Rui Yin and Brian G Pierce. Evaluation of alphafold antibody–antigen modeling with implications for improving predictive accuracy.Protein Science, 33(1):e4865, 2024
2024
-
[12]
Benchmarking alphafold for protein complex modeling reveals accuracy determinants.Protein Science, 31(8):e4379, 2022
Rui Yin, Brandon Y Feng, Amitabh Varshney, and Brian G Pierce. Benchmarking alphafold for protein complex modeling reveals accuracy determinants.Protein Science, 31(8):e4379, 2022
2022
-
[13]
Accurate de novo prediction of protein contact map by ultra-deep learning model.PLoS computational biology, 13(1):e1005324, 2017
Sheng Wang, Siqi Sun, Zhen Li, Renyu Zhang, and Jinbo Xu. Accurate de novo prediction of protein contact map by ultra-deep learning model.PLoS computational biology, 13(1):e1005324, 2017. 112
2017
-
[14]
Improved protein structure prediction using predicted interresidue orientations.Proceedings of the National Academy of Sciences, 117(3): 1496–1503, 2020
Jianyi Yang, Ivan Anishchenko, Hahnbeom Park, Zhenling Peng, Sergey Ovchin- nikov, and David Baker. Improved protein structure prediction using predicted interresidue orientations.Proceedings of the National Academy of Sciences, 117(3): 1496–1503, 2020
2020
-
[15]
Improved protein structure prediction using potentials from deep learning
Andrew W Senior, Richard Evans, John Jumper, James Kirkpatrick, Laurent Sifre, Tim Green, Chongli Qin, Augustin Žídek, Alexander WR Nelson, Alex Bridgland, et al. Improved protein structure prediction using potentials from deep learning. Nature, 577(7792):706–710, 2020
2020
-
[16]
A structural biology community assessment of alphafold2 applications.Nature Structural & Molecular Biology, 29(11):1056–1067, 2022
Mehmet Akdel, Douglas EV Pires, Eduard Porta Pardo, Jürgen Jänes, Arthur O Zalevsky, Bálint Mészáros, Patrick Bryant, Lydia L Good, Roman A Laskowski, Gabriele Pozzati, et al. A structural biology community assessment of alphafold2 applications.Nature Structural & Molecular Biology, 29(11):1056–1067, 2022
2022
-
[17]
Colabfold: making protein folding accessible to all
Milot Mirdita, Konstantin Schütze, Yoshitaka Moriwaki, Lim Heo, Sergey Ovchin- nikov, and Martin Steinegger. Colabfold: making protein folding accessible to all. Nature methods, 19(6):679–682, 2022
2022
-
[18]
Can alphafold2 predict the impact of missense mutations on structure?Nature structural & molecular biology, 29(1):1–2, 2022
Gwen R Buel and Kylie J Walters. Can alphafold2 predict the impact of missense mutations on structure?Nature structural & molecular biology, 29(1):1–2, 2022
2022
-
[19]
Accurate structure prediction of biomolecular interactions with alphafold 3
Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J Ballard, Joshua Bambrick, et al. Accurate structure prediction of biomolecular interactions with alphafold 3. Nature, 630(8016):493–500, 2024
2024
-
[20]
Critical assessment of methods of protein structure prediction (casp)—round xiv.Proteins: Structure, Function, and Bioinformatics, 89(12):1607–1617, 2021
Andriy Kryshtafovych, Torsten Schwede, Maya Topf, Krzysztof Fidelis, and John Moult. Critical assessment of methods of protein structure prediction (casp)—round xiv.Proteins: Structure, Function, and Bioinformatics, 89(12):1607–1617, 2021. 113
2021
-
[21]
A large-scale experiment to assess protein structure prediction methods.Proteins: Structure, Function, and Bioinformatics, 23(3):ii–iv, 1995
John Moult, Jan T Pedersen, Richard Judson, and Krzysztof Fidelis. A large-scale experiment to assess protein structure prediction methods.Proteins: Structure, Function, and Bioinformatics, 23(3):ii–iv, 1995
1995
-
[22]
Prottrans: Toward understanding the language of life through self-supervised learn- ing.IEEE transactions on pattern analysis and machine intelligence, 44(10):7112–7127, 2021
Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rehawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steinegger, et al. Prottrans: Toward understanding the language of life through self-supervised learn- ing.IEEE transactions on pattern analysis and machine intelligence, 44(10):7112–7127, 2021
2021
-
[23]
Single-sequence protein struc- ture prediction by integrating protein language models.Proceedings of the National Academy of Sciences, 121(13):e2308788121, 2024
Xiaoyang Jing, Fandi Wu, Xiao Luo, and Jinbo Xu. Single-sequence protein struc- ture prediction by integrating protein language models.Proceedings of the National Academy of Sciences, 121(13):e2308788121, 2024
2024
-
[24]
Evaluating protein transfer learning with tape
Roshan Rao, Nicholas Bhattacharya, Neil Thomas, Yan Duan, Xi Chen, John Canny, Pieter Abbeel, and Yun S Song. Evaluating protein transfer learning with tape. Advances in neural information processing systems, 32, 2019
2019
-
[25]
Transformer protein language models are unsupervised structure learners.BioRxiv, pages 2020–12, 2020
Roshan M Rao, Joshua Meier, Tom Sercu, Sergey Ovchinnikov, and Alexander Rives. Transformer protein language models are unsupervised structure learners.BioRxiv, pages 2020–12, 2020
2020
-
[26]
Large language models generate functional protein sequences across diverse families.Nature biotechnology, 41(8):1099–1106, 2023
Ali Madani, Ben Krause, Eric R Greene, Subu Subramanian, Benjamin P Mohr, James M Holton, Jose Luis Olmos Jr, Caiming Xiong, Zachary Z Sun, Richard Socher, et al. Large language models generate functional protein sequences across diverse families.Nature biotechnology, 41(8):1099–1106, 2023
2023
-
[27]
Protgpt2 is a deep unsupervised language model for protein design.Nature communications, 13(1):4348, 2022
Noelia Ferruz, Steffen Schmidt, and Birte Höcker. Protgpt2 is a deep unsupervised language model for protein design.Nature communications, 13(1):4348, 2022. 114
2022
-
[28]
V (d) j recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation
Xiying Chi, Yue Li, and Xiaoyan Qiu. V (d) j recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology, 160(3):233–247, 2020
2020
-
[29]
G. W. Litman, J. P . Rast, M. J. Shamblott, R. N. Haire, M. Hulst, W. Roess, R. T. Litman, K. R. Hinds-Frey, A. Zilch, and C. T. Amemiya. Phylogenetic diversification of immunoglobulin genes and the antibody repertoire.Molecular Biology and Evolution, 10(1):60–72, jan 1993. doi: 10.1093/oxfordjournals.molbev.a040000
-
[30]
Masamichi Muramatsu, Kazuo Kinoshita, Sidonia Fagarasan, Shuichi Yamada, Yoichi Shinkai, and Tasuku Honjo. Class switch recombination and hypermuta- tion require activation-induced cytidine deaminase (aid), a potential rna editing enzyme.Cell, 102(5):553–563, sep 2000. doi: 10.1016/S0092-8674(00)00078-7
-
[31]
Germinal centers.Annual review of immunology, 30:429–457, 2012
Gabriel D Victora and Michel C Nussenzweig. Germinal centers.Annual review of immunology, 30:429–457, 2012
2012
-
[32]
V (d) j recombination: mechanism, errors, and fidelity.Mobile DNA III, pages 311–324, 2015
David B Roth. V (d) j recombination: mechanism, errors, and fidelity.Mobile DNA III, pages 311–324, 2015
2015
-
[33]
Computational strategies for dissecting the high- dimensional complexity of adaptive immune repertoires.Frontiers in immunology, 9: 224, 2018
Enkelejda Miho, Alexander Yermanos, Cédric R Weber, Christoph T Berger, Sai T Reddy, and Victor Greiff. Computational strategies for dissecting the high- dimensional complexity of adaptive immune repertoires.Frontiers in immunology, 9: 224, 2018
2018
-
[34]
Protein data bank: the single global archive for 3d macro- molecular structure data.Nucleic Acids Research, 47(D1):D520–D528, 2019
wwPDB Consortium. Protein data bank: the single global archive for 3d macro- molecular structure data.Nucleic Acids Research, 47(D1):D520–D528, 2019
2019
-
[35]
Sabdab: the structural antibody database
James Dunbar, Konrad Krawczyk, Jinwoo Leem, Terry Baker, Angelika Fuchs, Guy Georges, Jiye Shi, and Charlotte M Deane. Sabdab: the structural antibody database. Nucleic acids research, 42(D1):D1140–D1146, 2014. 115
2014
-
[36]
H3-opt: Accurate prediction of cdr-h3 loop structures of antibodies with deep learning.bioRxiv, pages 2023–08, 2023
Hedi Chen, Xiaoyu Fan, Shuqian Zhu, Yuchan Pei, Xiaochun Zhang, Xiaonan Zhang, Lihang Liu, Feng Qian, and Boxue Tian. H3-opt: Accurate prediction of cdr-h3 loop structures of antibodies with deep learning.bioRxiv, pages 2023–08, 2023
2023
-
[37]
Fast, ac- curate antibody structure prediction from deep learning on massive set of natural antibodies.Nature communications, 14(1):2389, 2023
Jeffrey A Ruffolo, Lee-Shin Chu, Sai Pooja Mahajan, and Jeffrey J Gray. Fast, ac- curate antibody structure prediction from deep learning on massive set of natural antibodies.Nature communications, 14(1):2389, 2023
2023
-
[38]
Accurate prediction of antibody function and structure using bio-inspired antibody language model.Briefings in Bioinformatics, 25 (4), 2024
Hongtai Jing, Zhengtao Gao, Sheng Xu, Tao Shen, Zhangzhi Peng, Shwai He, Tao You, Shuang Ye, Wei Lin, and Siqi Sun. Accurate prediction of antibody function and structure using bio-inspired antibody language model.Briefings in Bioinformatics, 25 (4), 2024
2024
-
[39]
The h3 loop of antibodies shows unique structural characteristics.Proteins: Structure, Function, and Bioinformatics, 85(7):1311–1318, 2017
Cristian Regep, Guy Georges, Jiye Shi, Bojana Popovic, and Charlotte M Deane. The h3 loop of antibodies shows unique structural characteristics.Proteins: Structure, Function, and Bioinformatics, 85(7):1311–1318, 2017
2017
-
[40]
A new clustering of antibody cdr loop conformations.Journal of molecular biology, 406(2):228–256, 2011
Benjamin North, Axel Lehmann, and Roland L Dunbrack Jr. A new clustering of antibody cdr loop conformations.Journal of molecular biology, 406(2):228–256, 2011
2011
-
[41]
What does alphafold3 learn about antigen and nanobody docking, and what remains unsolved?bioRxiv, pages 2024–09, 2025
Fatima N Hitawala and Jeffrey J Gray. What does alphafold3 learn about antigen and nanobody docking, and what remains unsolved?bioRxiv, pages 2024–09, 2025
2024
-
[42]
Enhanced antibody- antigen structure prediction from molecular docking using alphafold2.Scientific Reports, 13(1):15107, 2023
Francis Gaudreault, Christopher R Corbeil, and Traian Sulea. Enhanced antibody- antigen structure prediction from molecular docking using alphafold2.Scientific Reports, 13(1):15107, 2023
2023
-
[43]
Reliable protein–protein docking with alphafold, rosetta, and replica exchange.Elife, 13:RP94029, 2025
Ameya Harmalkar, Sergey Lyskov, and Jeffrey J Gray. Reliable protein–protein docking with alphafold, rosetta, and replica exchange.Elife, 13:RP94029, 2025
2025
-
[44]
Three-dimensional structure of antibodies.Annual review of immunology, 6(1):555–580, 1988
Pedro M Alzari, Marie-Bénédicte Lascombe, and Roberto J Poljak. Three-dimensional structure of antibodies.Annual review of immunology, 6(1):555–580, 1988. 116
1988
-
[45]
Conformations of immunoglobulin hypervariable regions.Nature, 342(6252): 877–883, 1989
Cyrus Chothia, Arthur M Lesk, Anna Tramontano, Michael Levitt, Sandra J Smith- Gill, Gillian Air, Steven Sheriff, Eduardo A Padlan, David Davies, William R Tulip, et al. Conformations of immunoglobulin hypervariable regions.Nature, 342(6252): 877–883, 1989
1989
-
[46]
Canonical structures for the hypervariable regions of immunoglobulins.Journal of molecular biology, 196(4):901–917, 1987
Cyrus Chothia and Arthur M Lesk. Canonical structures for the hypervariable regions of immunoglobulins.Journal of molecular biology, 196(4):901–917, 1987
1987
-
[47]
The origin of cdr h3 structural diversity.Structure, 23(2):302–311, 2015
Brian D Weitzner, Roland L Dunbrack, and Jeffrey J Gray. The origin of cdr h3 structural diversity.Structure, 23(2):302–311, 2015
2015
-
[48]
Thiru Ramaraj, Timothy Angel, Edward A Dratz, and Sanjay Bhattacharyya. Antigen– antibody interface properties: composition, residue interactions, and features of 53 non-redundant structures.Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1824(3):520–532, 2012
2012
-
[49]
DIANE publishing, 1992
Elvin A Kabat.Sequences of proteins of immunological interest, volume 1. DIANE publishing, 1992
1992
-
[50]
Standard conformations for the canonical structures of immunoglobulins.Journal of molecular biology, 273(4): 927–948, 1997
Bissan Al-Lazikani, Arthur M Lesk, and Cyrus Chothia. Standard conformations for the canonical structures of immunoglobulins.Journal of molecular biology, 273(4): 927–948, 1997
1997
-
[51]
Imgt unique numbering for immunoglobulin and t cell receptor variable domains and ig su- perfamily v-like domains.Developmental & Comparative Immunology, 27(1):55–77, 2003
Marie-Paule Lefranc, Christelle Pommie, Manuel Ruiz, Veronique Giudicelli, Elise Foulquier, Lisa Truong, Valerie Thouvenin-Contet, and Gerard Lefranc. Imgt unique numbering for immunoglobulin and t cell receptor variable domains and ig su- perfamily v-like domains.Developmental & Comparative Immunology, 27(1):55–77, 2003
2003
-
[52]
James Dunbar and Charlotte M Deane. Anarci: antigen receptor numbering and re- 117 ceptor classification.Bioinformatics, 32(2):298–300, 2016. doi: 10.1093/bioinformatics/ btv552
-
[53]
50 years of antibody num- bering schemes: a statistical and structural evaluation reveals key differences and limitations.Antibodies, 13(4):99, 2024
Zirui Zhu, Katherine S Olson, and Thomas J Magliery. 50 years of antibody num- bering schemes: a statistical and structural evaluation reveals key differences and limitations.Antibodies, 13(4):99, 2024
2024
-
[54]
Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions.Proceedings of the National Academy of Sciences, 73(10):3628–3632, 1976
Nobumichi Hozumi and Susumu Tonegawa. Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions.Proceedings of the National Academy of Sciences, 73(10):3628–3632, 1976
1976
-
[55]
Targeting of somatic hypermutation.Nature Reviews Immunology, 6(8):573–583, 2006
Valerie H Odegard and David G Schatz. Targeting of somatic hypermutation.Nature Reviews Immunology, 6(8):573–583, 2006
2006
-
[56]
Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires.The Journal of Immunology, 201 (8):2502–2509, 2018
Aleksandr Kovaltsuk, Jinwoo Leem, Sebastian Kelm, James Snowden, Charlotte M Deane, and Konrad Krawczyk. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires.The Journal of Immunology, 201 (8):2502–2509, 2018
2018
-
[57]
Matthew I. J. Raybould, Aleksandr Kovaltsuk, Claire Marks, and Charlotte M. Deane. Cov-abdab: the coronavirus antibody database.Bioinformatics, 37(5):734–735, mar
-
[58]
doi: 10.1093/bioinformatics/btaa739
-
[59]
Attention is all you need.Advances in Neural Information Processing Systems, 30, 2017
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Łukasz Kaiser, and Illia Polosukhin. Attention is all you need.Advances in Neural Information Processing Systems, 30, 2017
2017
-
[60]
Long short-term memory.Neural compu- tation, 9(8):1735–1780, 1997
Sepp Hochreiter and Jürgen Schmidhuber. Long short-term memory.Neural compu- tation, 9(8):1735–1780, 1997
1997
-
[61]
Deep residual learning 118 for image recognition
Kaiming He, Xiangyu Zhang, Shaoqing Ren, and Jian Sun. Deep residual learning 118 for image recognition. InProceedings of the IEEE conference on computer vision and pattern recognition, pages 770–778, 2016
2016
-
[62]
Jimmy Lei Ba, Jamie Ryan Kiros, and Geoffrey E Hinton. Layer normalization.arXiv preprint arXiv:1607.06450, 2016
work page internal anchor Pith review Pith/arXiv arXiv 2016
-
[63]
Self-supervised learning: Generative or contrastive.IEEE transactions on knowledge and data engineering, 35(1):857–876, 2021
Xiao Liu, Fanjin Zhang, Zhenyu Hou, Li Mian, Zhaoyu Wang, Jing Zhang, and Jie Tang. Self-supervised learning: Generative or contrastive.IEEE transactions on knowledge and data engineering, 35(1):857–876, 2021
2021
-
[64]
BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding
Jacob Devlin, Ming-Wei Chang, Kenton Lee, and Kristina Toutanova. Bert: Pre- training of deep bidirectional transformers for language understanding.arXiv preprint arXiv:1810.04805, 2019
work page internal anchor Pith review Pith/arXiv arXiv 2019
-
[65]
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proceedings of the National Academy of Sciences, 118(15):e2016239118, 2021
2021
-
[66]
Representation learning: A review and new perspectives.IEEE transactions on pattern analysis and machine intelligence, 35(8):1798–1828, 2013
Yoshua Bengio, Aaron Courville, and Pascal Vincent. Representation learning: A review and new perspectives.IEEE transactions on pattern analysis and machine intelligence, 35(8):1798–1828, 2013
2013
-
[67]
Direct-coupling analysis of residue coevolution captures native contacts across many protein families.Proceedings of the National Academy of Sciences, 108(49):E1293–E1301, 2011
Faruck Morcos, Andrea Pagnani, Bryan Lunt, Arianna Bertolino, Debora S Marks, Chris Sander, Riccardo Zecchina, José N Onuchic, Terence Hwa, and Martin Weigt. Direct-coupling analysis of residue coevolution captures native contacts across many protein families.Proceedings of the National Academy of Sciences, 108(49):E1293–E1301, 2011
2011
-
[68]
Protein 3d structure computed from evolutionary sequence variation.PloS one, 6(12):e28766, 2011
Debora S Marks, Lucy J Colwell, Robert Sheridan, Thomas A Hopf, Andrea Pag- 119 nani, Riccardo Zecchina, and Chris Sander. Protein 3d structure computed from evolutionary sequence variation.PloS one, 6(12):e28766, 2011
2011
-
[69]
High-resolution de novo structure prediction from primary sequence.BioRxiv, pages 2022–07, 2022
Ruidong Wu, Fan Ding, Rui Wang, Rui Shen, Xiwen Zhang, Shitong Luo, Chenpeng Su, Zuofan Wu, Qi Xie, Bonnie Berger, et al. High-resolution de novo structure prediction from primary sequence.BioRxiv, pages 2022–07, 2022
2022
-
[70]
Se (3)-transformers: 3d roto-translation equivariant attention networks
Fabian Fuchs, Daniel Worrall, Volker Fischer, and Max Welling. Se (3)-transformers: 3d roto-translation equivariant attention networks. InAdvances in Neural Information Processing Systems, volume 33, pages 1970–1981, 2020
1970
-
[71]
E (n) equivariant graph neural networks
Vıctor Garcia Satorras, Emiel Hoogeboom, and Max Welling. E (n) equivariant graph neural networks. InInternational conference on machine learning, pages 9323–9332. PMLR, 2021
2021
-
[72]
Equivariant graph neural networks for 3d macromolecular structure
Bowen Jing, Stephan Eismann, Patricia Suriana, Raphael JL Townshend, and Ron Dror. Equivariant graph neural networks for 3d macromolecular structure. InICLR 2021 Workshop on Geometrical and Topological Representation Learning, 2021
2021
-
[73]
Mihaly Varadi, Stephen Anyango, Mandar Deshpande, Sreenath Nair, Cindy Natas- sia, Galabina Yordanova, David Yuan, Oana Stroe, Galen Wood, Agata Laydon, et al. Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models.Nucleic acids research, 50(D1): D419–D427, 2022
2022
-
[74]
Scoring function for automated assessment of protein structure template quality.Proteins: Structure, Function, and Bioinformatics, 57 (4):702–710, 2004
Yang Zhang and Jeffrey Skolnick. Scoring function for automated assessment of protein structure template quality.Proteins: Structure, Function, and Bioinformatics, 57 (4):702–710, 2004
2004
-
[75]
Tm-align: a protein structure alignment algorithm based on the tm-score.Nucleic Acids Research, 33(7):2302–2309, 2005
Yang Zhang and Jeffrey Skolnick. Tm-align: a protein structure alignment algorithm based on the tm-score.Nucleic Acids Research, 33(7):2302–2309, 2005. 120
2005
-
[76]
Dockq: a quality measure for protein-protein docking models.PloS one, 11(8):e0161879, 2016
Sankar Basu and Björn Wallner. Dockq: a quality measure for protein-protein docking models.PloS one, 11(8):e0161879, 2016
2016
-
[77]
Xiaomin Fang, Fan Wang, Lihang Liu, Jingzhou He, Dayong Lin, Yingfei Xiang, Xiaonan Zhang, Hua Wu, Hui Li, Le Song, et al. Helixfold-single: Msa-free protein structure prediction by using protein language model as an alternative.Research Square, 2022. doi: 10.21203/rs.3.rs-1969991/v1. Preprint
-
[78]
Language models enable zero-shot prediction of the effects of mutations on protein function.Advances in neural information processing systems, 34:29287–29303, 2021
Joshua Meier, Roshan Rao, Robert Verkuil, Jason Liu, Tom Sercu, and Alex Rives. Language models enable zero-shot prediction of the effects of mutations on protein function.Advances in neural information processing systems, 34:29287–29303, 2021
2021
-
[79]
Single-sequence protein structure prediction using a lan- guage model and deep learning.Nature Biotechnology, 40(11):1617–1623, 2022
Ratul Chowdhury, Nazim Bouatta, Surojit Biswas, Christina Floristean, Anant Kharkar, Koushik Roy, Charlotte Rochereau, Gustaf Ahdritz, Joanna Zhang, George M Church, et al. Single-sequence protein structure prediction using a lan- guage model and deep learning.Nature Biotechnology, 40(11):1617–1623, 2022
2022
-
[80]
Openfold: Retraining alphafold2 yields new insights into its learn- ing mechanisms and capacity for generalization.Nature Methods, 21:1514–1524, 2024
Gustaf Ahdritz, Nazim Bouatta, Christina Floristean, Sachin Kadyan, Qinghui Xia, William Gerecke, Timothy J O’Donnell, Daniel Berenberg, Ian Fisk, Niccolò Zanichelli, et al. Openfold: Retraining alphafold2 yields new insights into its learn- ing mechanisms and capacity for generalization.Nature Methods, 21:1514–1524, 2024
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.