Genetically Aligned Patient Representations Improve Hematological Diagnosis
Pith reviewed 2026-06-29 08:19 UTC · model grok-4.3
The pith
A patient encoder trained to align white blood cell images with karyotype and mutation data improves hematological diagnostic tasks over slide-level foundation models.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
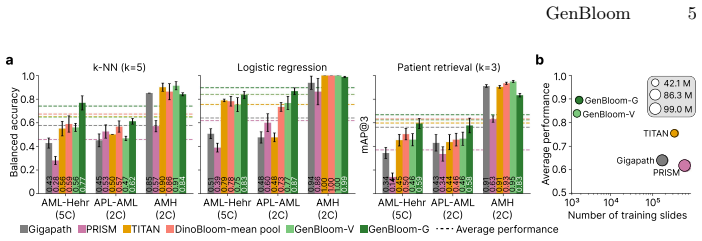
The genetically aligned patient encoder, built by first pretraining a vision transformer aggregator with an iBOT head on a large patient cohort and then applying supervised contrastive loss on acute myeloid leukemia cases to match single-cell images to karyotype and somatic mutation data, produces representations that improve performance on hematological diagnostic tasks and enable off-the-shelf retrieval of diseases and genetic alterations.
What carries the argument
The two-stage genetically aligned patient encoder that uses supervised contrastive loss on AML patients to align visual features with karyotype and mutation data after iBOT pretraining.
If this is right
- The aligned encoder outperforms slide-level histopathology foundation models on hematological diagnostic tasks.
- The model supplies off-the-shelf retrieval of diseases and genetic alterations from image queries.
- Incorporating genetic data raises the quality of the patient representations.
- The framework matches existing clinical diagnostic workflows that combine cytology with cytogenetics and molecular genetics.
Where Pith is reading between the lines
- The same alignment strategy could be tested on other paired image-genetic datasets outside hematology to check whether the two-stage recipe generalizes.
- Retrieval performance might allow construction of image-based search tools that return both diagnosis and expected genetic profile in one step.
- If the alignment holds on broader cohorts, future models could infer likely genetic alterations directly from images and flag cases needing confirmatory genetic testing.
Load-bearing premise
The supervised contrastive loss trained only on acute myeloid leukemia patients produces visual-genetic alignments that transfer to diagnostic tasks across the full range of hematological conditions.
What would settle it
An experiment in which the genetically aligned encoder shows no accuracy gain over the vision-only pretrained baseline on a held-out test set of non-AML hematological diagnoses.
Figures
read the original abstract
Multimodal alignment of histopathology encoders with transcriptomic and genomic data has been shown to significantly improve performance in downstream diagnostic tasks. Hematological cytology is unique in that visual single-cell evaluation is often paired with cytogenetics and molecular genetics for blood cancer diagnosis. In this study, we present a framework to align single white blood cell images with chromosomal aberrations (karyotype) and somatic mutations from targeted gene panels. Our training strategy follows a two-stage approach: (i) self-supervised, vision-only pretraining of a transformer aggregator using an iBOT head on a cohort of over 1500 patients, and (ii) genetic alignment via supervised contrastive loss on acute myeloid leukemia patients. Our genetically aligned patient encoder improves hematological diagnostic tasks, outperforming slide-level histopathology foundation models. Additionally, the model provides off-the-shelf retrieval capabilities for diseases and genetic alterations. Incorporating genetic data into patient encoders increases the quality of patient representations, providing a framework that aligns with clinical diagnostic workflows and paves the way for future multimodal hematology-specific AI. The code and model weights are available at https://github.com/marrlab/GenBloom.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a two-stage framework for aligning single white blood cell images with genetic data (karyotypes and somatic mutations) in hematological cytology. Stage (i) performs self-supervised pretraining of a vision transformer aggregator using an iBOT head on a cohort of >1500 patients; stage (ii) applies supervised contrastive loss exclusively on AML patients to align image embeddings with genetic labels. The central claim is that the resulting genetically aligned patient encoder improves performance on hematological diagnostic tasks relative to slide-level histopathology foundation models, while also enabling off-the-shelf retrieval of diseases and genetic alterations. Code and model weights are released.
Significance. If the empirical claims hold after addressing generalization, the work would be significant for multimodal representation learning in medical imaging. It provides a concrete method to incorporate routinely available cytogenetic and molecular labels into visual patient encoders in a manner consistent with clinical hematology workflows. The open release of code and weights strengthens reproducibility and enables follow-on research.
major comments (2)
- [Abstract] Abstract: the claim that the genetically aligned encoder 'improves hematological diagnostic tasks' and 'outperforms slide-level histopathology foundation models' is stated without any quantitative metrics, baseline comparisons, statistical significance tests, or dataset splits. This absence makes it impossible to assess the magnitude or reliability of the reported improvement.
- [Training strategy description] Training strategy description (and results sections): the supervised contrastive alignment stage uses genetic positives/negatives derived exclusively from AML patients. The headline claim requires that this AML-specific alignment produces embeddings that improve diagnostic performance across broader hematological tasks (including non-AML). No cross-disease ablation isolating the contribution of the contrastive stage on held-out non-AML cohorts is reported; without such evidence the generalization of the learned embedding space remains unsubstantiated.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and constructive feedback on our manuscript. We address each of the major comments below, providing clarifications and indicating the revisions we plan to implement.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that the genetically aligned encoder 'improves hematological diagnostic tasks' and 'outperforms slide-level histopathology foundation models' is stated without any quantitative metrics, baseline comparisons, statistical significance tests, or dataset splits. This absence makes it impossible to assess the magnitude or reliability of the reported improvement.
Authors: We agree that the abstract presents a high-level overview without specific quantitative details. The results section of the manuscript includes detailed performance metrics, baseline comparisons (including slide-level histopathology foundation models), statistical significance tests, and information on dataset splits. To address this concern and make the abstract more informative, we will revise it to include key quantitative results, such as the magnitude of improvement and relevant p-values, subject to space constraints. revision: yes
-
Referee: [Training strategy description] Training strategy description (and results sections): the supervised contrastive alignment stage uses genetic positives/negatives derived exclusively from AML patients. The headline claim requires that this AML-specific alignment produces embeddings that improve diagnostic performance across broader hematological tasks (including non-AML). No cross-disease ablation isolating the contribution of the contrastive stage on held-out non-AML cohorts is reported; without such evidence the generalization of the learned embedding space remains unsubstantiated.
Authors: The self-supervised pretraining is performed on a large cohort of over 1500 patients encompassing multiple hematological conditions, establishing a general visual representation. The supervised contrastive alignment leverages genetic data available primarily from AML patients. Our experiments evaluate the resulting encoder on a range of hematological diagnostic tasks, including those involving non-AML cases. We acknowledge the value of an explicit ablation study. We will add a cross-disease analysis in the revised manuscript to better isolate the contribution of the contrastive alignment stage, including performance on held-out non-AML cohorts where possible. Note that comprehensive genetic labels are more readily available for AML, which informs the design. revision: partial
Circularity Check
No significant circularity; derivation is self-contained
full rationale
The paper's core claim rests on a standard two-stage pipeline: self-supervised iBOT pretraining on >1500 patients followed by supervised contrastive alignment using external karyotype and mutation labels from AML cases. Neither stage reduces to its inputs by construction, nor does any equation or result rename a fitted quantity as a prediction. No load-bearing self-citations, uniqueness theorems, or ansatzes imported from prior author work appear in the abstract or described training strategy. The approach therefore remains externally falsifiable against held-out diagnostic performance and does not exhibit any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Bioinformatics35(24), 5365–5366 (2019)
Abrams, Z.B., Zhang, L., Abruzzo, L.V., Heerema, N.A., Li, S., Dillon, T., Ro- driguez, R., Coombes, K.R., Payne, P.R.: Cytogps: a web-enabled karyotype anal- ysis tool for cytogenetics. Bioinformatics35(24), 5365–5366 (2019)
2019
-
[2]
Leukemia pp
Dasdelen, M.F., Kukuljan, I., Lienemann, P., Ozlugedik, F., Sadafi, A., Hehr, M., Spiekermann, K., Pohlkamp, C., Marr, C.: AI-based hematological malignancy prediction from peripheral blood smears in a large diagnostic laboratory cohort. Leukemia pp. 1–5 (2026)
2026
-
[3]
Nature medicine pp
Ding, T., Wagner, S.J., Song, A.H., Chen, R.J., Lu, M.Y., Zhang, A., Vaidya, A.J., Jaume,G.,Shaban,M.,Kim,A.,etal.:Amultimodalwhole-slidefoundationmodel for pathology. Nature medicine pp. 1–13 (2025)
2025
-
[4]
An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale
Dosovitskiy, A., Beyer, L., Kolesnikov, A., Weissenborn, D., Zhai, X., Unterthiner, T., Dehghani, M., Minderer, M., Heigold, G., Gelly, S., et al.: An image is worth 16x16 words: Transformers for image recognition at scale. arXiv preprint arXiv:2010.11929 (2020)
work page internal anchor Pith review Pith/arXiv arXiv 2010
-
[5]
Leukemia36(1), 111–118 (2022)
Eckardt, J.N., Middeke, J.M., Riechert, S., Schmittmann, T., Sulaiman, A.S., Kramer, M., Sockel, K., Kroschinsky, F., Schuler, U., Schetelig, J., et al.: Deep learning detects acute myeloid leukemia and predicts npm1 mutation status from bone marrow smears. Leukemia36(1), 111–118 (2022)
2022
-
[6]
Leukemia36(7), 1939–1942 (2022) 10 Dasdelen et al
Fuhrmann, I., Lenk, M., Haferlach, T., Stengel, A., Hutter, S., Baer, C., Meggen- dorfer, M., Kern, W., Haferlach, C.: Aml, nos and aml-mrc as defined by multilin- eage dysplasia share a common mutation pattern which is distinct from aml-mrc as defined by mds-related cytogenetics. Leukemia36(7), 1939–1942 (2022) 10 Dasdelen et al
1939
-
[7]
PLOS Digital Health2(3), e0000187 (2023)
Hehr, M., Sadafi, A., Matek, C., Lienemann, P., Pohlkamp, C., Haferlach, T., Spiekermann, K., Marr, C.: Explainable ai identifies diagnostic cells of genetic aml subtypes. PLOS Digital Health2(3), e0000187 (2023)
2023
-
[8]
Jaffe, E.S.: Pathology and genetics of tumours of haematopoietic and lymphoid tissues, vol. 3. Iarc (2001)
2001
-
[9]
In: Advances in Neural Information Processing Systems (NeurIPS)
Khosla, P., Teterwak, P., Wang, C., Sarna, A., Tian, Y., Isola, P., Maschinot, A., Liu, C., Krishnan, D.: Supervised contrastive learning. In: Advances in Neural Information Processing Systems (NeurIPS). vol. 33, pp. 15871–15882 (2020)
2020
-
[10]
leukemia36(7), 1703–1719 (2022)
Khoury, J.D., Solary, E., Abla, O., Akkari, Y., Alaggio, R., Apperley, J.F., Be- jar, R., Berti, E., Busque, L., Chan, J.K., et al.: The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and his- tiocytic/dendritic neoplasms. leukemia36(7), 1703–1719 (2022)
2022
-
[11]
In: International Conference on Medical Image Computing and Computer-Assisted Intervention
Koch, V., Wagner, S.J., Kazeminia, S., Sancar, E., Hehr, M., Schnabel, J.A., Peng, T., Marr, C.: Dinobloom: a foundation model for generalizable cell embeddings in hematology. In: International Conference on Medical Image Computing and Computer-Assisted Intervention. pp. 520–530. Springer (2024)
2024
-
[12]
Reprint of’Cytogenetic and Genome Research 2020, Vol
McGowan-Jordan, J., Hastings, R.J., Moore, S.: Iscn 2020: An International Sys- tem for Human Cytogenomic Nomenclature (2020). Reprint of’Cytogenetic and Genome Research 2020, Vol. 160, No. 7-8’. Karger Medical and Scientific Publish- ers (2020)
2020
-
[13]
DINOv2: Learning Robust Visual Features without Supervision
Oquab, M., Darcet, T., Moutakanni, T., Vo, H., Szafraniec, M., Khalidov, V., Fernandez, P., Haziza, D., Massa, F., El-Nouby, A., et al.: Dinov2: Learning robust visual features without supervision. arXiv preprint arXiv:2304.07193 (2023)
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[14]
Leukemia16(1), 53–59 (2002)
Schoch, C., Schnittger, S., Bursch, S., Gerstner, D., Hochhaus, A., Berger, U., Hehlmann, R., Hiddemann, W., Haferlach, T.: Comparison of chromosome banding analysis, interphase-and hypermetaphase-fish, qualitative and quantitative pcr for diagnosis and for follow-up in chronic myeloid leukemia: a study on 350 cases. Leukemia16(1), 53–59 (2002)
2002
-
[15]
arXiv preprint arXiv:2405.10254 (2024) 12 J
Shaikovski, G., Casson, A., Severson, K., Zimmermann, E., Wang, Y.K., Kunz, J.D., Retamero, J.A., Oakley, G., Klimstra, D., Kanan, C., et al.: Prism: A multi- modal generative foundation model for slide-level histopathology. arXiv preprint arXiv:2405.10254 (2024)
-
[16]
NPJ precision oncology5(1), 38 (2021)
Sidhom, J.W., Siddarthan, I.J., Lai, B.S., Luo, A., Hambley, B.C., Bynum, J., Duffield, A.S., Streiff, M.B., Moliterno, A.R., Imus, P., et al.: Deep learning for diagnosis of acute promyelocytic leukemia via recognition of genomically imprinted morphologic features. NPJ precision oncology5(1), 38 (2021)
2021
-
[17]
Molecular-driven foundation model for oncologic pathology.arXiv preprint arXiv:2501.16652, 2025
Vaidya, A., Zhang, A., Jaume, G., Song, A.H., Ding, T., Wagner, S.J., Lu, M.Y., Doucet, P., Robertson, H., Almagro-Perez, C., et al.: Molecular-driven foundation model for oncologic pathology. arXiv preprint arXiv:2501.16652 (2025)
-
[18]
Blood, The Journal of the American Society of Hematology100(7), 2292–2302 (2002)
Vardiman, J.W., Harris, N.L., Brunning, R.D.: The world health organization (who) classification of the myeloid neoplasms. Blood, The Journal of the American Society of Hematology100(7), 2292–2302 (2002)
2002
-
[19]
PLOS Computational Biology22(2), e1013950 (2026)
Wang, W., Zhang, X., Xiong, Y.: Transcriptomic-guided whole-slide image classi- fication for molecular subtype identification. PLOS Computational Biology22(2), e1013950 (2026)
2026
-
[20]
Nature630(8015), 181–188 (2024)
Xu, H., Usuyama, N., Bagga, J., Zhang, S., Rao, R., Naumann, T., Wong, C., Gero, Z., González, J., Gu, Y., et al.: A whole-slide foundation model for digital pathology from real-world data. Nature630(8015), 181–188 (2024)
2024
-
[21]
iBOT: Image BERT Pre-Training with Online Tokenizer
Zhou, J., Wei, C., Wang, H., Shen, W., Xie, C., Yuille, A., Kong, T.: ibot: Image bert pre-training with online tokenizer. arXiv preprint arXiv:2111.07832 (2021)
work page internal anchor Pith review Pith/arXiv arXiv 2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.